
Abstract
A single mouse click on the topic tumor necrosis factor (TNF) in PubMed reveals about 50 000 articles providing one or the other information about this pleiotropic cytokine or its relatives. This demonstrates the enormous scientific and clinical interest in elucidating the biology of a molecule (or rather a large family of molecules), which began now almost 30 years ago with the description of a cytokine able to exert antitumoral effects in mouse models. Although our understanding of the multiple functions of TNF in vivo and of the respective underlying mechanisms at a cellular and molecular level has made enormous progress since then, new aspects are steadily uncovered and it appears that still much needs to be learned before we can conclude that we have a full comprehension of TNF biology. This review shortly covers some general aspects of this fascinating molecule and then concentrates on the molecular mechanisms of TNF signal transduction. In particular, the multiple facets of crosstalk between the various signalling pathways engaged by TNF will be addressed.
Similar content being viewed by others

General aspects of TNF biology
The principle of an antitumoral response of the immune system in vivo has been recognized already about 100 years ago by the physician William B Coley. About 30 years ago, a soluble cytokine termed tumor necrosis factor (TNF) has been identified that is produced upon activation by the immune system, able to exert significant cytotoxicity on many tumor cell lines and to cause tumor necrosis in certain animal model systems. In 1984, the cDNA of TNF was cloned, the structural and functional homology to lymphotoxin (LT) α was realized, and several years later, two membrane receptors, each capable of binding both cytokines, were identified. Subsequently, it was recognized that TNF is the prototypic member of a large cytokine family, the TNF ligand family.
TNF is primarily produced as a type II transmembrane protein arranged in stable homotrimers (Figure 1).1,2 From this membrane-integrated form the soluble homotrimeric cytokine (sTNF) is released via proteolytic cleavage by the metalloprotease TNF alpha converting enzyme (TACE).3 The soluble 51 kDa trimeric sTNF tends to dissociate at concentrations below the nanomolar range, thereby losing its bioactivity. The 17 kDa TNF protomers are composed of two antiparallel β-pleated sheets with antiparallel β-strands, forming a ‘jelly roll’ β-structure, typical for the TNF ligand family, but also found in viral capsid proteins.4

Membrane-bound TNF (memTNF) and soluble TNF (sTNF) derived thereof, both bind to two members of the TNF-receptor superfamily, TNF-R1 and TNF-R2. While memTNF activates both TNF receptors, sTNF predominantly stimulates TNF-R1 and has limited signalling capacities on TNF-R2
The members of the TNF ligand family exert their biological functions via interaction with their cognate membrane receptors, comprising the TNF receptor (TNF-R) family.5 The members of the TNF-R family contain one to six cysteine-rich repeats in their extracellular domain, typically each with three cysteine bridges.6 Two receptors, TNF-R1 (TNF receptor type 1; CD120a; p55/60) and TNF-R2 (TNF receptor type 2; CD120b; p75/80) bind membrane-integrated TNF (memTNF) as well as soluble TNF (sTNF), but also the secreted homotrimeric molecule lymphotoxin-α (LTα). The functional role of LTα in man is largely undefined and will not be discussed in this review. TNF-R1 and TNF-R2 each contain four cysteine-rich repeats in their extracellular domains and form elongated shapes, which interact with the lateral grooves of the trimeric ligand formed between each two of its three protomers.6,7 Ligand-dependent trimerization of the receptors was long considered as the key event for signal initiation. However, initial receptor activation now appears more complicated, because the distal cysteine-rich domains of TNF-R1 and TNF-R2 mediate homophilic interaction of receptor molecules in the absence of ligand. These preligand binding assembly domains (PLAD)8 may therefore keep receptors in a silent, homomultimerized status and antagonize spontaneous autoactivation, the latter being frequently observed upon overexpression. Accordingly, ligand binding to the preformed TNF-R complex either induces an activating conformational change of an a priori signal competent receptor complex or it allows the formation of higher-order receptor complexes, which acquire signal competence.
TNF-R1 is constitutively expressed in most tissues, whereas expression of TNF-R2 is highly regulated and is typically found in cells of the immune system. In the vast majority of cells, TNF-R1 appears to be the key mediator of TNF signalling, whereas in the lymphoid system TNF-R2 seems to play a major role. Generally, the importance of TNF-R2 is likely to be underestimated, because this receptor can only be fully activated by memTNF, but not sTNF.9 The cause for this difference is not fully understood yet, but the different stabilities, that is half-lifes, of the individual ligand/receptor complexes may contribute to this.9,10 The extracellular domains of both receptors can be proteolytically cleaved, yielding soluble receptor fragments with potential neutralizing capacity.11 Owing the lack of cooperativity in ligand binding, however, the affinities of soluble receptors are low compared to their membrane-integrated forms. TNF neutralizing agents for clinical use that were constructed on the basis of the soluble receptors have therefore been engineered as dimeric IgG fusion proteins.12 Like TNF, TNF-R2 is cleaved by TACE.13 The processing enzyme(s) responsible for TNF-R1 cleavage is still undefined, but TNF-R1 cleavage is obviously an important step in the regulation of cellular TNF responsiveness, as cleavage-resistant TNF-R1 mutations are linked with dominantly inherited autoinflammatory syndromes (TNF-R1-associated periodic syndromes; TRAPS).14
The intracellular domains of TNF-R1 and TNF-R2 that do not possess any enzymatic activity define them as representatives of the two main subgroups of the TNF-R family, the death domain-containing receptors and the TRAF-interacting receptors, respectively. TNF-R1 contains a protein–protein interaction domain, called death domain (DD).15 The DD can recruit other DD-containing proteins and couples the death receptors to caspase activation and apoptosis.16 In addition, as described in detail below, TNF-R1 is also a potent activator of gene expression via indirect recruitment of members of the TNF receptor-associated factor (TRAF) family. TNF-R2 directly recruits TRAF2, induces gene expression and intensively crosstalks with TNF-R1.
TNF is mainly produced by macrophages, but also by a broad variety of other tissues including lymphoid cells, mast cells, endothelial cells, fibroblasts and neuronal tissue. Large amounts of sTNF are released in response to lipopolysaccharide and other bacterial products. In concert with other cytokines, TNF is considered to be a key player in the development of septic shock.17 Whereas high concentrations of TNF induce shock-like symptoms, the prolonged exposure to low concentrations of TNF can result in a wasting syndrome, that is, cachexia. This can be found for example in tumor patients. Indeed, the biological mediator of cachexia, originally described in an animal model of trypanosoma infection and thus called cachectin, has been later unravelled as TNF.18
TNF exerts an extreme spectrum of bioactivities and most cells show at least some TNF responsiveness. In general, TNF may be considered to represent a major proinflammatory mediator, with an optional capacity to induce apoptosis. In (patho)physiological situations, TNF shows a remarkable functional duality, being strongly engaged both in tissue regeneration/expansion and destruction. One important example is the role of TNF in neurodegeneration. CNS-specific overexpression of TNF in transgenic mice revealed infiltrating CD4+ and CD8+ T cells, astrocytosis, microgliosis and demyelination.19 Although from these transgenic animal models, TNF and TNF-R signalling has been implicated as important for the onset of demyelinating disease, TNF must also be recognized as a reactive cytokine that is upregulated in response to traumatic and excitotoxic injury of the brain, thus potentially exerting protective functions. Of interest, TNF receptors can have counteracting functions, at least in neuronal tissues, as recently demonstrated in a murine model of retinal ischemia, where TNF-R1 apparently aggravated tissue destruction, whereas TNF-R2 was protective via activation of PKB/Akt.20 In a different transgenic model, a targeted AU repeat deletion of 69 bp in the 3′untranslated region of the TNF gene resulted in an enhanced stability of TNF mRNA and elevated protein level in fibroblasts, but not in a significant change in TNF responses to LPS challenge. These animals develop clinical signs of arthritis and colitis.21 However, when this modified TNF transgene is crossed into TNF-R2-deficient mice, a near to normal phenotype is observed, pointing to a significant contribution of this receptor for the development of chronic inflammatory diseases.21 A further example for the two-edged role of TNF in vivo is liver regeneration after partial hepatectomy. In TNF-R1-deficient animals, hepatocyte DNA synthesis is severely impaired, indicating that TNF signalling through TNF-R1 is involved in liver regeneration.22 In contrast, in models of acute hepatotoxicity TNF acting via TNF-R1 appears as a key player in liver destruction.23 Owing to its strong proinflammatory and immunostimulatory activities, TNF is, in general, an important mediator of progression of many autoimmune diseases. Important examples are rheumatoid arthritis and inflammatory bowel disease (Crohn's disease), where significant clinical improvement can be achieved when patients are treated with TNF neutralizing agents.24,25 Thus, the question as to whether TNF contributes to or protects from tissue damage in acute or chronic diseases is probably wrongly posed. The accumulating data rather suggest a very differential TNF action and indicate that tissue type, precise cellular context and TNF-R composition, timing and duration of TNF action are important parameters determining the net effect of TNF action in vivo.
The developmental role of TNF and its receptors has also been addressed by gene targeting approaches. TNF, LTα and LTβ have been inactivated either alone or in combinations and the respective receptors (TNF-R1, TNF-R2, LTβR) were targeted as well. Knockout mice lacking LTβR, the receptor for a heterotrimeric ligand consisting of LTα and LTβ, are devoid of all lymph nodes, Peyer's patches and gut-associated lymphatic tissue, providing clear evidence for the essential role of this receptor in secondary lymphoid tissue development. Interestingly, TNF−/− and TNF-R1−/− mice share some features of the LTβR−/− phenotype, but also reveal unique characteristics, thus defining both redundant and nonredundant functions for each of these molecules in lymph node formation.26,27
Further, in full accordance with the important role of TNF as a mediator of the innate immune system, impaired defense against certain intracellular pathogens is observed in TNF-R1- and TNF-deficient animals, whereas parameters of the adaptive immune system like CD8+ T-cell cytotoxicity, mixed lymphocyte responses, T-cell-independent B-cell response and most parameters of T-cell-dependent B-cell response remain grossly normal.28 TNF- or TNF-R1-deficient mice show enhanced sensitivity when challenged with, for example, Mycobacterium tuberculosis,29 Lysteria monocytogenes30 or Leishmannia major.31 In mycobacterial infections, especially the formation of granuloma is TNF dependent.32 TNF-R1- and TNF-deficient mice, but not TNF-R2 knockout animals, die from a fulminant necrotizing encephalitis when orally infected with a low-virulent strain of Toxoplasma gondii.33 However, in other infection models, TNF knockout mice show delayed pathological reactions when challenged with pathogens. This has been observed for example in rabies virus infection,34 the acute phase of infection by Yersinia enterocolitica,35 and a model of cerebral malaria.36 Independent from its role in host defense, TNF might also play a role in downregulating the immune system after a successful response.37 Together, these examples clearly show that the specific role of TNF in infection is highly dependent on the type of the pathogen, the general context and stage of the infection. Very recent data from patients with inflammatory bowel disease, who have been treated Qwith the TNF-R2–IgG fusion protein etanercept to antagonize TNF activity, support a crucial role of TNF in defense against intracellular pathogens: during treatment an exacerbation of Mycobacterium tuberculosis enteritis was observed.38
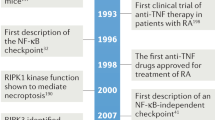
The first described and thus the name giving action of TNF was its antitumoral activity in mouse tumor models.39 As a result of TNFs strong cytotoxic activity on some tumor cells in vitro,40 TNF was initially considered as a widely applicable, direct tumoricidal reagent. However, meanwhile it is evident that TNF-mediated tumor rejection in vivo is dependent on a functional immune response and most likely independent of TNF's capability to induce directly apoptosis in tumor target cells.41 Moreover, systemic TNF application in humans is limited by severe side effects, ranging from influenza-like symptoms to the development of life-threatening symptoms of shock.17 Nevertheless, more recent data show that TNF can be successfully applied as a tumor therapeutic under conditions that prevent systemic TNF action. Thus, high concentrations of TNF in combination with the chemotherapeutic drug melphalan, applied under isolated limb perfusion conditions, yielded superior response rates and limb salvage in metastatic sarcoma.42 The underlying mechanism involves destruction of the tumor vasculature leading to a necrotic destruction of the tumor.43 For a successful application of TNF in other tumor entities future developments need to accommodate these results, aiming at genetically engineered TNF-based constructs that display site-specific action.
TNF-induced activation of NF-κB
Nuclear factor kappa B (NF-κB) comprises a group of dimeric transcription factors consisting of various members of the NF-κB/Rel family. NF-κB proteins are involved in the transcriptional activation of a huge number of inflammatory-related genes in response to cytokines, for example, TNF and IL-1, bacterial products and some forms of physical ‘stress’, for example, UV radiation or reactive oxygen species.44 In the recent years, it became also evident that NF-κB induces a variety of antiapoptotic factors, which is of importance for the regulation of TNF-R1-mediated triggering of the apoptotic machinery of the cell (see below). In mammalian cells, five members of the NF-κB/Rel family are known: NF-κB1/p50, which is constitutively processed from its precursor p105 by proteolysis, NF-κB2/p52, which is inducibly processed from its precursor p100, c-Rel, RelA/p65 and RelB.45 The NF-κB/Rel proteins share the conserved Rel homology domain (RHD), which mediates dimerization, DNA binding, nuclear localization and interaction with members of the I-κB protein family – the inhibitory counterparts of the NF-κB/Rel proteins.45 The I-κB family is characterized by six or seven ankyrin repeats and includes I-κBα, I-κBβ, I-κBγ, I-κBɛ, Bcl-3 and the precursors of NF-κB1 and NF-κB2, p105 and p100, respectively, that contain seven carboxy-terminal ankyrin repeats in addition to their amino-terminal RHD. I-κBα, I-κBβ and I-κBɛ also contain an amino-terminal regulatory domain that allows stimulus-induced degradation of these proteins.46 When functions of NF-κB or I-κB are described in the following paragraphs, in most cases, we refer to data obtained with p65/p50 heterodimers and the I-κBα isoform.
In an uninduced state, cellular I-κB proteins interact with NF-κB dimers to mask their nuclear location sequence, thereby retaining the ternary complex of NF-κB and I-κB in the cytoplasm. TNF, like a variety of other inducers, can stimulate proteolytic degradation of I-κB by the proteasome, thus liberating NF-κB and allowing nuclear translocation.46 For full activation, NF-κB must be further modified by phosphorylation of its subunits. Several kinases including mitogen activated protein kinases (MAPK) and protein kinase C (PKC) isoforms have been implicated in this secondary modification of NF-κB activity (see below). The level at which the various I-κB degradation-inducing signalling pathways converge is the activation of a multicomponent protein kinase complex, the I-κB kinase (IKK) complex. The activated IKK complex is able to phosphorylate the regulatory domain of I-κB and this marks it for recognition by an SKP1-Cullin-Fbox-type E3 ubiquitin–protein ligase complex. The IKK complex is believed to comprise a heteromer of two related I-κB kinases, called IKK1 and IKK2 (IKKα and IKKβ),47,48,49 the regulatory protein NEMO (Fip-3, IKKγ, IKKAP)50,51,52,53,54 and a homodimer of the heat shock protein-90 (Hsp90), as well as two or three molecules of the Hsp90-asscociated cdc37 protein.55 NEMO-deficient cells are completely impaired in NF-κB activation by all inducers investigated so far56,57,58 suggesting that this noncatalytic component has an essential role in the IKK complex. Analyses of IKK1- and IKK2-deficient mouse embryonic fibroblasts revealed that both I-κB kinases are nonredundant in their function and contribute differently to TNF-induced NF-κB activation (Table 1). IKK2-deficient mice are strongly impaired in TNF-induced I-κB phosphorylation, but nevertheless show residual DNA binding of NF-κB, significant production of NF-κB-driven genes and almost unchanged p65 phosphorylation after TNF stimulation.59,60 In contrast, TNF-induced I-κB phosphorylation and DNA binding of NF-κB were found to be unaffected in IKK1 deficient mouse embryonic fibroblasts in two studies,61,62 but another study reported significant reduction in TNF-induced NF-κB binding and inhibition of NF-κB target genes.63 There is also evidence that IKK1, but not IKK2, has a role in a second pathway leading to the activation of NF-κB2 by promoting its processing from the p100 precursor. Interestingly, this pathway is independent of I-κB degradation,64,65 However, there is yet no evidence that this pathway plays a role in TNF signalling. In good agreement with an essential role of the IKK complex in TNF-induced NF-κB activation is that all effects of this response are completely abrogated in mouse embryonic fibroblasts (MEFs) of IKK1–IKK2 double-deficient mice.66 In particular, this indicates that the recently identified IKK-related kinases TBK/T2K/NAK67,68,69 and IKKɛ70 cannot substitute for IKK1 and IKK2 in TNF-signalling.
The initial event in TNF-induced activation of the IKK complex is ligand-induced reorganization of preassembled TNF-R1 complexes (Figure 1). The intrinsic property of the death domain of TNF-R1 to self-aggregate and therefore to signal independent of ligand is masked in preassembled TNF-R1 complexes by binding of the silencer of death domain (SODD) protein.71 After ligand binding, SODD dissociates from TNF-R1 complexes and the death domain-containing adaptor protein TRADD is recruited to the death domain of TNF-R1 by homophilic interactions of the death domains.72 TNF-R1-bound TRADD then serves as an assembly platform for binding of TNF receptor-associated factor (TRAF) 2 and the death domain-containing serine–threonine kinase RIP (receptor-interacting kinase).73 TRAF2 is a member of the phylogenetically conserved TRAF protein family.74 The characteristic feature of the TRAF proteins is a carboxy-terminal homology domain of about 180 aa, the TRAF domain, which mediates a wide range of protein–protein interactions including binding to MAP3 kinases, various regulators, and non-death domain-containing members of the TNF receptor family.74 With the exception of TRAF1, all TRAF proteins have an amino-terminal RING finger, which is followed by five or seven evenly spaced zinc fingers.74 The association of TRAF2 to TNF-R1-bound TRADD is mediated by the interaction of its carboxy-terminal TRAF domain with the amino-terminal death domain of TRADD.75 In contrast, RIP is recruited to the DD of TNF-R1-bound TRADD via its carboxy-terminal death domain.73 RIP is also able to interact with TRAF2 via its amino-terminal kinase domain and its central intermediate domain.73 However, studies with TRAF2- and RIP- deficient mouse embryonic fibroblasts have shown that both molecules can be independently recruited into the TNF-R1 signalling complex.76 Moreover, these studies showed that TRAF2 is sufficient to recruit the IKK complex into the TNF-R1 signalling complex whereas RIP is necessary for the activation of the IKKs.76 Although RIP is able to interact with NEMO51,77 in the yeast two-hybrid system, studies with RIP-deficient cells showed that this interaction is dispensable for recruitment of the IKK complex to TNF-R1.76 Thus, a minimal model of a TNF-induced NF-κB activation comprises TRAF2, acting as a receptor proximal adaptor (via TRADD binding) that recruits the IKK complex to the TNF-R1 signalling complex, thereby enabling RIP to activate the kinases of the IKK complex (Figure 2). However, there are several lines of evidence that the interplay of TRAF2, RIP and the IKK complex is more complicated. In RIP-deficient cells, similar amounts of IKK1 and IKK2 are found in the TNF-R1 signalling complex compared to wt cells, but the amount of coprecipitated NEMO is significantly reduced.78 These data, together with the finding that increasing amounts of TRAF2 interfere with the interaction of IKK1 and NEMO,78 suggest that TRAF2 binding weakens the coherence of the IKK complex. Remarkably, RIP is able to compensate the TRAF2 inhibitory effect on the IKK1/2–NEMO interaction78 and possibly stabilizes the IKK complex after TRAF2-mediated recruitment to the TNF-R1 signalling complex. A puzzling detail in this regard is the observation that in RIP-deficient cells a significantly increased amount of TRAF2 and TRADD is recruited to TNF-R1 after TNF treatment76,78 without an effect on the recruitment of IKK1/2.78 Overexpression studies73 and reconstitution experiments in RIP-deficient cells76,79 suggest that the kinase activity of RIP is dispensable for IKK activation. It is conceivable that RIP activates the IKKs indirectly via mitogen-activated protein kinase kinase kinase MEKK3, as RIP can interact with MEKK3, and MEKK3-deficient mouse embryonic fibroblasts show strongly reduced NF-κB activation in response to TNF and IL1.80 However, marginal TNF-induced NF-κB activation has been reported for MEKK3−/− mouse embryonic fibroblasts pointing to the possibility that other kinases can substitute to some extent for MEKK3.80 Indeed, a possible candidate is MEKK1, which is able to interact with TRAF2 as well as with RIP after TNF stimulation in human HEK293 cells and is activated by TNF in a RIP-dependent manner in the human T-cell line Jurkat.81,82 Inconsistent with a role of MEKK1 in TNF-induced NF-κB activation are findings showing that this response is normal in MEKK1-deficient embryonal stem cells as well as in fibroblasts and macrophages derived from MEKK1-deficient mice.83,84 However, it cannot be ruled out that this discrepancy is based on cell- or species-specific distinct roles of various MAP3K in TNF signalling.

Scheme of TNF-R1-induced NF-κB activation. Components of the NF-κB pathway that can be cleaved by caspases during apoptosis are shown in gray boxes
Although TRAF2 has been mainly recognized as a physical link between the TNF-R1 signalling complex and the IKK complex it seems possible that this molecule has additional functions in NF-κB activation. Indeed, the TRAF2-related TRAF6 molecule is able to interact with PKCζ, an atypical protein kinase C,85 which has also been implicated in TNF-induced NF-κB activation.86 Analyses of cells derived from PKCζ-deficient mice point to a cell type-specific and potentially multi-functional role in TNF-mediated NF-κB activation (Table 1). While in lung tissue of PKCζ-deficient mice IKK activation by TNF is impaired, embryonal fibroblasts from the same animals show IKK activation similar to WT cells.86 Nevertheless, DNA binding of NF-κB and transcription of an NF-κB-driven luciferase reporter gene have been found to be significantly reduced also in this cell type. A possible explanation for these observations could be the capability of PKCζ to phosphorylate p65 in its RHD, which is responsible for DNA binding.86,87 In this regard, PKCζ resembles the catalytic subunit of protein kinase A (cPKA), which can be released from an I-κB/NF–κB/cPKA complex in response to LPS and which is involved in NF-κB activation by phosphorylation of RHD serine 276.88 In this case, phosphorylation of serine 276 is necessary to allow recruitment of the transcriptional coactivator CPB/p300. Interaction of CPB/p300 and p65 is mediated by two motifs within p65, one within the RHD comprising phosphorylated serine 276 and one unphosphorylated site in the C-terminal region of p65, which is only accessible upon phosphorylation of serine 276.88 The role of PKCζ-dependent phosphorylation of p65 would then be clearly distinct from the action of some other kinases, which stimulate NF-κB activity by phosphorylation of p65 in its carboxy-terminal transactivation domain without affecting DNA binding (see below). The cell type-restricted NF-κB deficiency in knockout cells could be because of the functional redundancy of PKCζ and the closely related atypical PKCλ, which display different expression patterns. Indeed, PKCζ as well as PKCλ interact with the IKK complex after TNF stimulation89 and both kinases indirectly interact with RIP via the adaptor protein p62.90 The complex role of PKCζ exemplifies that activation of the IKK complex, degradation of I-κB, nuclear translocation of p65 and DNA binding may not suffice for efficient transcription of NF-κB responsive genes. To obtain full NF-κB function, additional stimulation of the transactivation potential of p65 by phosphorylation at serines 529 and 536 appears to be required. This can be accomplished by constitutively active kinases like casein kinase II, which is able to phosphorylate serine 529 of p65 after its release from I-κB,91 or by the serine–threonine kinase Akt upon TNF stimulation via an IKK1-dependent pathway.92 TNF can stimulate Akt in a cell type-specific manner via the phosphoinositide-3OH kinase (PI3K) pathway.93,94,95,96,97,98,99,100,101 In agreement with a role of the PI3K/Akt pathway in TNF-induced NF-κB activation, it has been found that the dual specificity phosphatase PTEN, which dephosphorylates and inactivates phosphatidylinositol 3-phosphate, inhibits TNF-induced transcription of NF-κB-driven genes.102,103 However, although these studies partly used the same experimental models, there are considerable discrepancies regarding the steps in NF-κB activation that are affected by PTEN-dependent inhibition of Akt. Two recent studies using IKK1- and IKK2- deficient mouse embryonic fibroblasts reported different results concerning the role of IKK1 and IKK2 in Akt-mediated transactivation of the p65 subunit of NF-κB.92,104 In both studies, IKK2 was necessary for Akt-dependent p65 transactivation, whereby in one study myristoylated and therefore constitutively active Akt (Myr-Akt) and in the other study TNF was used to induce transactivation of p65.92,104 However, in the study of Sizemore et al.104 TNF-induced transactivation was in addition dependent on IKK1 and the Akt pathway, whereas overexpressed Myr-Akt was able to transactivate p65 in IKK1-deficent cells.104 In agreement with an important role of IKK1 in TNF-induced transactivation of p65, another report using IKK1-deficient mouse embryonic fibroblasts shows that TNF-induced upregulation of the endogenous NF-κB target genes I-κBα, IL6 and M-CSF is impaired in these cells.63
Another level of complexity in TNF-induced NF-κB activation became apparent from the analyses of mice deficient for poly(ADP-ribose) polymerase 1 (PARP-1; see Table 1), a nuclear DNA repair enzyme activated by DNA strand breaks.105 Unexpectedly, PARP-1−/− mice were highly resistant to LPS-induced endotoxic shock because of an impaired NF-κB response towards the inflammatory mediators LPS and TNF.106,107 While degradation of I-κB and translocation of NF-κB were normal in PARP-1-deficient cells, DNA-binding and transcriptional activation were found to be severely reduced.106,107 Subsequent in vitro studies have then shown that PARP-1 interacts with both p65 and p50 by two independent domains.108,109,110 However, there are contradictory data concerning the importance of the PARP-1 enzymatic activity for NF-κB activation.108,109,111,112,113 TNF is able to activate PARP-1 via the production of ROS, which in turn causes PARP-inducing DNA damage. However, it is an open question whether TNF-induced PARP-1 activation via ROS is a prerequisite for its role in TNF-induced NF-κB activation or whether PARP-1 exerts its NF-κB-supporting capability independent of prior activation. Interestingly, TNF-induced PARP-1 activation by the production of ROS and subsequent DNA damage has also been implicated in the regulation of the balance between apoptosis and necrosis (see below). It will be interesting to see whether the roles of PARP-1 in NF-κB activation and in cell death induction are related in some way.
Remarkably, mouse embryonic fibroblasts from mice deficient for GSK3β or TBK/T2K/NAK, two kinases previously not thought to be involved in TNF signalling, also exert reduced NF-κB-dependent transcription, but normal nuclear translocation and DNA binding in response to TNF, suggesting a role of these kinases in p65 transactivation, also.68,114 Kinase-inactive mutants of TBK/T2K/NAK failed to interfere with TNF-induced NF-κB activation, thus the role of TBK/T2K/NAK in TNF-dependent NF-κB activation might be rather of structural nature than implying its enzymatic capabilities.67,69 Nevertheless, a more complex, maybe multifunctional, role of TBK/T2K/NAK in NF-κB activation is possible as this kinase is able to phosphorylate IKK269 and its kinase activity is necessary for NF-κB activation by overexpression of TANK.67 However, the molecular mechanisms underlying the effects of GSK3ß and TBK/T2K/NAK on TNF-induced NF-κB activation are not defined yet.
While the pathways leading from TNF/TNF-R1 interaction to activation of the IKK complex and NF-κB are comparably well understood, the mechanisms involved in the termination of the TNF-induced NF-κB response are rather unclear. There is evidence that TNF-selective, but also rather globally acting feedback mechanisms are utilized to terminate TNF-induced NF-κB activation. Complexes of soluble TNF and TNF-R1 are rapidly internalized115,116 opening the possibility that degradation in secondary lysosomes contributes to termination of TNF responses. By contrast, TNF-induced internalization seems to be required for efficient stimulation of some (JNK, aSMase, apoptosis), whereas other signalling pathways (nSMase, ERK, NF-κB) initiated by TNF-R1 are independent of internalization.117
NF-κB-dependent upregulation of NF-κB inhibitory proteins is another powerful mechanism involved in feedback inhibition of the NF-κB pathway. In particular, I-κBα and A20 have been identified as NF-κB inducible genes that are required for the postinduction repression of TNF-induced NF-κB activation.118,119,120 As I-κBα targets free p65/p50 subunits, it inhibits NF-κB activation by a variety of stimuli. In contrast, analyses of mouse embryonic fibroblasts of A20-deficient fibroblasts point to a more selective role. Mouse embryonic fibroblasts deficient for A20 show persistent activation of the IKK complex and prolonged DNA binding of NF-κB in response to TNF, whereas termination of the IL1-induced NF-κB activation remains unaffected.121 In contrast to the TNF-selective effects observed in A20-deficient cells, biochemical and transient overexpression studies point to a more general regulatory role of A20 affecting various MAP3K associated with NF-κB activation.122,123,124 A20 can interact with TRAF2120 as well as NEMO.77 In agreement with the IKK inhibitory role of A20 and its recruitment to the TNF-R1–IKK signalling complex it has been found that the capacity of the IKK complex, precipitated from whole cell lysates, to phosphorylate I-κB is greater than that of the TNF-R1-associated IKK complex.77 Interestingly, A20 promotes the phosphorylation of IKK1/2 in the context of the TNF-R1–IKK signalling complex.77 IKK2 undergoes progressive autophosphorylation at multiple serine residues in its carboxy-terminal end, thereby decreasing its kinase activity.125 It is therefore tempting to speculate that A20 stabilizes or promotes this autoinhibitory state of IKK2.
Stimulation of TNF-R1 can lead to a strong activation of the apoptotic pathway, in particular when protein synthesis is globally reduced or when the NF-κB pathway is compromised. Therefore, the fact that the NF-κB pathway targets, among others, a variety of antiapoptotic genes is of special interest. Indeed, there is growing evidence that the NF-κB and the apoptotic pathway are tightly connected. This aspect of TNF signalling is therefore discussed below in the context of TNF-induced apoptosis.
TNF-induced activation of JNK and p38-MAPK
TNF regularily induces the activation of kinases of the stress-activated protein kinase (SAPK)/c-Jun N-terminal kinase (JNK) group.126 The JNK isoforms are distantly related to mitogen-activated protein kinases (ERKs) and, like ERKs, are activated by dual phosphorylation on tyrosine and threonine residues. Upon activation, JNK kinases translocate into the nucleus and enhance the transcriptional activity of transcription factors, for example, c-Jun and ATF2, by phosphorylation of their amino-terminal activation domains.127 c-Jun belongs to a group of basic region-leucine zipper proteins that dimerize to form transcription factors commonly designated as activator protein-1 (AP-1).127 However, JNK kinases have also functions not related to c-Jun phosphorylation. The AP-1 proteins have an important role in a variety of cellular processes including proliferation, differentiation and induction, as well as prevention of apoptosis.127 Although TNF-induced JNK activation and c-Jun phosphorylation have been implicated in upregulation of collagenases,128 the chemoattractant MCP-1,129 E-selectin130 and in the regenerative response to liver injury,131,132 the importance of JNK activation for TNF-mediated cellular responses is otherwise poorly understood.
TNF-induced activation of the JNK pathway occurs through a nonapoptotic TRAF2-dependent pathway (Figure 3).133,134,135,136,137,138 It is evident from knockout mice and mice overexpressing a dominant-negative form of TRAF2 that this adaptor is necessary for coupling the JNK pathway to TNF-R1.137,138 Analyses of MKK7- and MKK4- deficient mouse embryonic fibroblasts suggest that MKK7 is essentially involved in TNF-induced JNK activation, whereas MKK4 contributes to optimal TNF-mediated JNK activation, but is not sufficient to evoke this response alone.139 The distinct roles of MKK7 and MKK4 in TNF-induced JNK activation most likely reflect the different, but complementary substrate specificities of these kinases. MKK7 preferentially phosphorylates threonine 180 of JNK,140 whereas MKK4 mainly phosphorylates tyrosine182.139 In this regard, it has been shown that MKK7 is able to activate WT JNK, but to a lesser degree also mutated JNK harboring a Tyr182Phe substitution, whereas MKK4 is only able to activate wt JNK.139 Therefore, TNF-induced activation of JNK seems not to depend completely on dual JNK phosphorylation. Thus, phosphorylation of threonine 180 by MKK7 could be sufficient to activate JNK in response to TNF and MKK4 could enhance TNF–MKK7-mediated JNK activation by phosphorylation of tyrosine 182. Remarkably, TNF activates MKK7 but not MKK4,141 suggesting that the basal activity of MKK4 is sufficient to allow maximal activation of JNK in response to TNF. While knockout mice have clearly proven the pivotal roles of TRAF2 and MKK7 for TNF-R1-mediated JNK activation, the nature and function of the MAP3K, which must fill the gap between TRAF2 and MKK7, has not been satisfactorily identified yet. Based on their ability to interact with TRAF2, the JNK-inducing MAP3 kinases MEKK1 and ASK1 potentially have a role in TNF-induced JNK activation,81,142,143 but studies in mice deficient for these kinases could not support an essential role in TNF-induced JNK activation.84,144 However, as discussed in detail later, a role of ASK1 in prolonged TNF-induced JNK activation, which occurs under apoptotic conditions, is supported by the ASK1 knockout mice. It is also possible that a role of ASK1 and/or MEKK1 in TNF-induced JNK activation is masked by redundancies. Indeed, biochemical data indicate that several parallel pathways link TNF-R1/TRAF2 to MKK7 and JNK.

Scheme of TNF-R1-induced activation of p38–MAPK and JNK. JNK activation by TNF can occur by caspase-dependent and/or caspase-independent pathways. The latter is based on ‘reactive oxygen species’-dependent and/or GCK-dependent activation of the JNK cascade module, whereby both are regulated by TRAF2. TNF-R1-mediated, caspase-dependent activation of JNK takes place as a consequence of apoptosis induction and is therefore dependent on the TRADD–FADD–caspase-8 axis. The poorly defined connection between RIP and MKK3 is indicated by a dotted line
There is evidence that TRAF2 mediates TNF-induced JNK activation through interaction with members of the germinal center kinase (GSK) family (Figure 3). The GCK family comprises a group of serine–threonine kinases homologous to the yeast Ste20p kinase that can be subdivided into two groups according to the structure of their carboxy-terminal regulatory domain. The carboxy-terminal regulatory domain of group I GCKs contains two or more PEST motifs, binding sites for SH3-domains and a highly conserved stretch of about 350 aa comprising a leucine-rich amino-terminal part and a so-called C-terminal region.145 Group I GCKs act as proximal activators of MAPK pathways by phosphorylation of MAP3Ks.145 Group II GCKs are homologous in their amino-terminal kinase domain to group I GCKs but differ drastically from these molecules in the architecture of the carboxy-terminal regulatory domain. In particular, GCKs of the group II do not stimulate any of the currently known MAPK pathways.145 Several group I GCKs (germinal center kinase (GCK),146 GCK-like kinase (GLK),147 GCK-related (GCKR),148 HPK/GCK-like kinase (HGK),149 NIK-like embryo-specific kinase (NESK),150 TRAF2 and Nck-interacting kinase (TNIK)151) have been implicated in TNF-mediated JNK activation because of the following findings: they become activated by TNF (NESK, HGK, GCKR, GLK), interact with TRAF2 (TNIK, GCK, GCKR152) and dominant-negative mutants of these kinases interfere with TNF-induced JNK activation (NESK, HGK, GCKR). The group I GCKs seem to channel stimulation of TNF-R1 to the JNK cascade by concomitant interaction with TRAF2 and MEKK1 and phosphorylation-dependent activation of the latter.146,153 GCK-induced MEKK1 activation correlated with the enhancement of the oligomerization of MEKK1,153 a process, which on its own is sufficient to drive the activation of MEKK1-dependent signalling pathways.81,153 The suggested role of group I GCKs in TNF-mediated JNK activation is mainly based on correlation (TNF/TRAF2 activates GCKs – GCKs activate JNK) or transient overexpression experiments with dominant-negative mutants, which are difficult to interpret in terms of causal relations, considering that related molecules may compete for common endogenous upstream or downstream signalling components. Additional studies, especially analyses of knockout mice, will therefore be necessary to figure out the relative (and maybe cell-type specific) contribution of the various GCKs to TNF-induced JNK activation.
A second GCK-independent pathway used by TNF-R1 to activate JNK is based on the production of reactive oxygen species (ROS) and activation of apoptosis signal-regulating kinase-1 (ASK1), a member of the MAP3K family (Figure 3).154 Stimulation of TNF-R1 can result in the TRAF2-dependent increase of ROS of mitochondrial origin by yet poorly understood mechanisms.155 Stimulation of TNF-R1 induces interaction of ASK1 with TRAF2 and leads to activation of ASK1 by antioxidant-sensitive mechanisms.156,157 In agreement with the latter, thioredoxin (Trx) has been identified as an ASK1 interacting protein.157 Trx contains a redox-active center composed of two cysteine residues and can exist in an oxidized form containing a disulfide-bridge in the active center (Trx-S2) or in a reduced form with two free SH-groups (Trx-(SH)2). Trx can be oxidized by various ROS and thereby protects to some extent from the cytotoxic effects of TNF,158 which is partly based on the production of these molecules. Under reducing conditions, Trx exists in its Trx-(SH)2 form and is able to bind and inhibit ASK1.157 The generation of ROS leads to the oxidation of Trx-(SH)2 to Trx-S2, which is no longer able to interact with ASK1.157 In addition, it has been shown that TNF-induced ASK1 activation and TRAF2–ASK1 interaction require prior dissociation of Trx-ASK1 complexes.159 Thus, a model obtrudes in which TNF-induced generation of ROS leads to the oxidation of Trx, release of ASK1 from inhibitory Trx-ASK1 complexes and subsequent formation of JNK-inducing TRAF2–ASK1 complexes.159 Remarkably, TRAF2-dependent activation of ASK1 correlates with oligomerization of the kinase.156 Thus, both TRAF2-GCK-mediated and TRAF2–ASK1-mediated activation of JNK seem to involve oligomerization-dependent activation of MAP3Ks. TRAF2 has therefore a dual role in TNF-induced ASK1 activation, firstly as an inducer of Trx-ASK1 dissociation by ROS induction and secondly, as an activating oligomerization scaffold for ASK1. In agreement with the existence of parallel TRAF2-dependent, JNK-activating pathways (TRAF2–GCKs–MEKK1 versus TRAF2–ROS–ASK1), it has been found that Trx-(SH)2 completely blocks TRAF2–ASK1 interaction, but has only a partial inhibitory effect on TRAF2-mediated JNK activation.159
TNF not only robustly activates the JNK-inducing MAP kinase cascade, but also the p38-MAPK signalling cascade. Indeed, many aspects of TNF-induced JNK activation hold also true for TNF-induced activation of the p38–MAPK cascade (Figure 3). Both, JNK and p38, are transiently activated by TNF, but show prolonged activation under apoptotic conditions. Moreover, TRAF2, ASK1 and MEKK1, which have been implicated in TNF-induced JNK activation, are also strong inducers of the p38–MAPK pathway.146,154,160 Nevertheless, there is clear evidence that upstream and downstream of the MAP3 kinase level differences exist between JNK and p38 activation. For example, the various GCKs, which have been implicated in TNF-induced activation of JNK, are unable to stimulate the p38–MAPK cascade.147,149,150,151,161 In addition, it has been shown that a deletion mutant of RIP, lacking its intermediate domain, interferes with TRAF2-mediated activation of p38–MAPK, but failed to inhibit TRAF2-induced JNK activation.146 Thus, TNF may signal p38–MAPK activation via an axis comprising TRAF2 and RIP, whereas JNK activation occurs via TRAF2–GCKs–MEKK1/ASK1. In agreement with the latter, TRAF2 knockout mice are impaired in TNF-induced JNK activation,138 whereas RIP-deficient mice appear normal in this regard.162 Unfortunately, studies concerning activation of p38–MAPK in mice deficient for TRAF2 and/or RIP have not been published yet.
Mouse embryonic fibroblasts of MKK3-deficient mice show a strong reduction in TNF-induced activation of p38–MAPK, but no effect on TNF-induced JNK activation (Table 1).163 In agreement with the important role of the p38-MAPK signalling pathway as a mediator of inflammatory processes, TNF-induced production of IL1 and IL6 is almost completely blocked in MKK3−/− mouse embryonic fibroblasts. However, the exact mode of action of p38–MAPK in TNF-induced upregulation of IL1 and IL6 is not clear yet. p38–MAPK may act by activation of various transcription factors including ATF2, CHOP, CREB and ELK1164 but it could also enhance TNF-induced production of IL1 and IL6 by increasing the stability of the respective mRNAs via activation of MAP kinase-activated protein kinase 2.165,166 Remarkably, p38–MAPK has also been discussed as a mediator of NF-κB transactivation.167,168 To which extent the various effector mechanisms of p38–MAPK are of importance in TNF-induced production of inflammatory cytokines remains to be seen.
TRADD-independent signalling pathways
A number of proteins have been described to interact with TNF-R1 outside its death domain, some of them with undefined impact on TNF signals. The WD-repeat protein FAN (factor associated with neutral sphingomyelinase activation) has been identified by its binding capacity to the membrane-proximal region of TNF-R1.169 FAN interacts with one of its five WD-repeats with a nine AA stretch of TNF-R1 located directly in the membrane-proximal region of the DD and appears involved in the activation of the neutral sphingomyelinase (nSMase).169,170 Defects in cutaneous barrier repair have been found in mice with a genetic deletion of FAN.170 The role of nSMase-derived ceramide is largely undefined, especially as at least in some cells nSMase becomes only activated very transiently during only the first 3 min of TNF stimulation.171 A proapoptotic function of nSMase has been described in one report using a dominant-negative form of FAN,172 whereas ERK1/2 and phospholipase A2 activation are not linked to FAN.173 Other proteins binding TNF-R1 upstream the DD include a regulatory component of the 26S proteasome, called TRAP2, 55.11 or p97, that binds to aa residues 234–308 of TNF-R1, and which might be involved in TNF-mediated regulation of proteasomal functions.174,175,176 In addition, the mitochondrially localized Hsp75, also called TRAP1, has been found to bind TNF-R1 membrane proximal of the DD.177,178
TNF-induced activation of the classical MAP kinases, that is the ERKs, is, in most cells, absent or only moderate when compared to TNF activation of the stress-activated protein kinases179 (see above) or compared to activation of ERKs via mitogenic receptor tyrosine kinase (RTK) pathways.180 Rather, a negative feedback of TNF on ERK activation triggered by RTK signals has been observed in several cell lines.181 TNF-R2 can also induce JNK, but not ERKs.182 A protein containing a domain with low DD homology termed MADD or Rab3-GAP binds to the DD of TNF-R1 and is able to activate MAP kinase pathways.183,184 Various splice variants of MADD exist which are also termed DENN.185 Further, the adapter protein Grb2 binds to a PLAP motif of TNF-R1, thereby potentially linking this receptor via SOS to Ras, c-Raf and the ERKs.186 This signal, however, appears not sufficient to efficiently activate the MAP kinases, as FAN/nSMase-derived ceramide acting on the ceramide-activated protein kinase (CAP-K;187) is necessary to fully activate c-Raf.186 Consistent with these data, survival of osteoclasts by TNF is mediated by Akt and ERKs and can be blocked by inhibitors of the ERK-activating kinase MEK-1, but also by a peptide interfering with FAN/PLAP domain interaction.188
Molecular mechanisms of TNF-induced cell death
Like other death receptors, TNF-R1 is able to signal cell death via its cytoplasmic death domain in a variety of cell lines. However, in vivo TNF-induced apoptosis seems to have only a minor role compared to its overwhelming function in the regulation of inflammatory processes. Indeed, the high systemic toxicity of TNF is caused by cellular mediators like NO and not related to its apoptosis-inducing capability.189 Remarkably, mice deficient in p65 or other components involved in TNF-induced NF-κB activation are embryonally lethal or die early after birth because of massive TNF-dependent liver failure (Table 1). Thus, the death-inducing capability of TNF is masked in vivo by concomitant activation of NF-κB. Although other death receptors are also able to activate the NF-κB pathway, they show prominent apoptotic functions in vivo. Moreover, in some experimental situations Fas-mediated NF-κB activation becomes apparent only when parallel apoptosis induction is blocked.190 Thus, while in TNF-R1 signalling NF-κB activation dominates over apoptosisinduction, in Fas or TRAIL-R1/2 signalling apoptosis-induction is dominant over NF-κB activation. As outlined below in detail, the apoptotic and the NF-κB pathway are inhibitory to each other. Thus, there is a situation that both pathways are connected by self-amplifying inhibitory circuits that may be responsible for the predominant in vivo specification of TNF-R1 as an NF-κB-inducing receptor and of Fas as a death-inducing receptor.
There is genetic evidence from knock-out mice and mutagenized cell lines that all death receptors investigated so far critically depend on the death domain-containing adaptor protein FADD and caspase-8 and -10 to induce cell death (Table 1).191,192,193,194,195,196,197,198,199 In the case of Fas and the TRAIL death receptors, a death-inducing signalling complex (DISC) that contains FADD and caspase-8/10 has been defined by immunoprecipitation of the endogenous molecules. Studies with deletion mutants of Fas, FADD and caspase-8 show that activated Fas recruits FADD by homophilic interaction of the DD of these molecules.200 Receptor-bound FADD in turn interacts with caspase-8/10 via the death effector domains contained in the amino-terminal parts of both molecules.201,202 There is evidence that the Fas-DISC is a supramolecular complex of several trimeric or higher-order Fas complexes.203 Thus, FADD-mediated recruitment brings several caspase-8/10 molecules in close proximity and thereby facilitates autoproteolytic activation of these proteases. In contrast to Fas and TRAIL death receptors, TNF-R1 is indirectly linked to FADD, namely by TRADD which is also responsible for bridging TNF-R1 to TRAF2 and the IKK complex.75 However, while several groups were able to immunoprecipitate the TNF-R1-IKK signalling complex from TNF-treated cells (see above), a comparable demonstration of the TNF-R1-DISC was not successful yet. The lack of an immunoprecipitable TNF-R1-DISC could reflect a comparably low stability of the complex, but could also indicate that the efficient formation of such a complex requires special, yet poorly understood circumstances. In agreement with the existence of mechanisms that selectively regulate the formation and/or activity of the TNF-R1-DISC, several groups have observed that depletion of TRAF2, which is a major part of the TNF-R1 signalling complex, but does not or only modestly interact with Fas, sensitises cells for the apoptotic action of TNF,204,205,206,207,208,209,210 whereas Fas- and TRAIL-mediated cell death remain unaffected.205,210 A TNF-R1 selective apoptosis-regulating process could be the TRAF2-mediated recruitment of the antiapoptotic cIAP1 and cIAP2 proteins to TNF-R1.210,211 cIAP1 and the closely related cIAP2 protein have been originally identified as molecules present in the TNF-R2 signalling complex.212 Both are typical members of the inhibitor of apoptosis protein family,213 which bind and inhibit caspase-3 and -7 via their amino-terminal BIR (baculovirus IAP repeat) domains,214 a structural feature common to all IAP family members.213 In agreement with an antiapoptotic role of TRAF2-mediated recruitment of cIAP1 and cIAP2, it has been found that concerted overexpression of TRAF1, TRAF2, cIAP1 and cIAP2 efficiently interferes with TNF-R1-induced apoptosis and activation of caspase-8.215 So, it seems that in the TNF-R1 signalling complex TRAF2-bound cIAP1/2 molecules are able to block activation of caspase-8, which is independently recruited into the TNF-R1 signalling complex via the TRADD–FADD axis and which is otherwise no substrate for these IAP proteins.214 Depletion of cytoplasmic, therefore TNF-R1 accessible, TRAF2 can be induced by stimulation of non-death domain-containing members of the TNF receptor family able to recruit TRAF2 and to induce its subsequent proteasomal degradation.207,210 To destine TRAF2 for proteasomal degradation the molecule has to be ubiquitinated.207,209 Indeed, additional to their caspase inhibitory BIR domains the carboxy-terminal RING domain of cIAP1 and cIAP2 can act as E3 ubiquitin ligase216 involved in the proteasomal degradation of caspase-3 and –7,217 and cIAP1 and cIAP2 themselves.216 Although TRAF2 mediates recruitment of cAP1 and cIAP2 into the TNF-R2 signalling complex with comparable efficiencies,210 only cIAP1 seems to ubiquitinate TRAF2.207 In agreement with the hypotheses that TRAF2 depletion interferes with the formation of the caspase-8 inhibiting TNF-R1-TRAF/IAP complex mentioned above, it has been demonstrated that TNF-R2 stimulation enhances TNF-R1-induced caspase-8 activation.208,210 Hence, TNF-R2-dependent enhancement of TNF-R1-induced cell death appears to be based on two tightly linked mechanisms: First, on competition of the two TNF-Rs for binding of TRAF2 and the TRAF2-associated anti–apoptotic cIAP1 and cIAP2 proteins, and second, on the cIAP1-initiated degradation of TRAF2, which in turn enhances receptor competition for the remaining TRAF2, cIAP1 and cIAP2 molecules. Accordingly, cIAP1 would have an anti-apoptotic function upon recruitment into the TNF-R1 signalling complex, but would switch to a net proapoptotic function upon recruitment into the TNF-R2 signalling complex.
Downstream of caspase-8 processing TNF-R1-induced apoptosis occurs in principle via the same routes as described for other death receptors like Fas and TRAIL-R1/2 (see the parallel review articles). In brief, in type I cells active caspase-8 alone is sufficient to robustly induce caspase-3 activity and the execution phase of apoptosis (Figure 4). Thus, release of cytochrome c from mitochondria and subsequent formation of the caspase-3-inducing apoptosome complex are dispensable for the apoptotic process in this type of cells (see the parallel review articles). However, in type II cells caspase-8- mediated activation of caspase-3 is inefficient and the apoptotic process therefore depends on a mitochondria-dependent amplification loop (Figure 4). Small quantities of active caspase-8 produced in type II cells are sufficient to activate proteolytically the BH3 domain-containing Bcl2 family member Bid.218,219 The truncated carboxy-terminal fragment of Bid (tBid) generated this way translocates to the mitochondria and promotes release of cytochrome c218,219 and Smac/Diablo220,221 in a Bax/Bak-dependent manner.222 The release of cytochrome c from mitochondria into the cytosol allows the ATP-dependent formation of a caspase-3 activating ‘apoptosome’, consisting of cytochrome c itself, Apaf-1 and caspase-9.223 Complementary to the action of the caspase-3-inducing apoptosome, Smac/Diablo binds and antagonizes the caspase inhibitors xIAP, cIAP1 and cIAP2.220,221 Activated caspase-3 is able to activate caspase-8224,225,226 thereby creating a positive feedback loop.225,226 to the pivotal role of the mitochondrial cytochrome c release in type II cells, these type of cells can be experimentally distinguished from type I cells by overexpression of Bcl-2. While in type I cells apoptosis induction by death receptors is not sensitive towards Bcl-2 overexpression, this molecule protects type II cells from the apoptotic effects of death receptors.227 Since the amount of active caspase-8 seems to be critical to determine type I or type II responsiveness and TNF-R1-mediated caspase-8 processing is less efficient compared to Fas, it seems conceivable that the same cells could behave as a type I cell or a type II cell, depending on the death receptor subtype, that is Fas or TNF-R1, respectively.

TNF-R1 stimulates a death-inducing network. Necrosis-related relationships are shown by dotted lines, apoptosis-related relationships are depicted with solid lines. NF-κB-inducible inhibitors of the cell death network are highlighted with gray boxes. Note that the active caspase-3 (casp.-3*) cleaves and inhibits PARP, thereby blocking the necrotic response. Cathepsin D-mediated activation of caspase-3 occurs most likely indirectly by Bid cleavage and is therefore shown by a gray dashed line. The role of RIP in the production of ROS has not been experimentally addressed yet. Thus, this relation is deduced from the receptor-proximal position of RIP and the finding that RIP is essentially involved in death receptor-induced necrosis
cIAP1, cIAP2 and TRAF1 have been identified as NF-κB target genes.215,228,229 Thus, ablation of the NF-κB pathway may interfere with the action of the caspase-8 inhibitory TNF-R1-TRAF/IAP complex and therefore sensitises for the apoptotic action of TNF. In particular, this is in good accordance with the increased TNF sensitivity observed in various knock-out mice where TNF-induced NF-κB activation is compromised (Table 1). Remarkably, the NF-κB pathway targets also several other antiapoptotic factors, including cFLIP,230,231 IEX-1L,232 Bfl-1/A1,233,234,235 XIAP,236 Bcl-Xl 234,237,238 and the TRAIL decoy receptor TRAIL-R3.239 All or part of these genes are most likely involved in or responsible for the variety of global, non-TNF-R1 selective anti-apoptotic effects of NF-κB. Upregulation of NF-κB, dependent antiapoptotic genes seems not to be the only way by which TNF counteracts induction of apoptosis by itself or other death inducers. In accordance with the known anti-apoptotic properties of the PI3K/Akt pathway, caused by phosphorylation and inactivation of proteins involved in apoptosis,240,241,242 there is growing evidence that the stimulation of this pathway by TNF not only contributes to TNF-dependent NF-κB activation, but also independently mediates some additional antiapoptotic signals of TNF.98,100
While the NF-κB pathway negatively regulates the apoptotic program, ongoing apoptosis in turn interferes with the activation of NF-κB. This is because of the caspase-mediated cleavage of several of the components utilized by this pathway including RIP, 243,244 TRAF1, 245 I-κB, 246 IKK2, 247 HPK1, 248 NIK,249 Akt252 as well as p50 250 and p65 250,251 themselves. Noteworthy, in most cases caspase-mediated cleavage results in release of fragments that can act in a dominant-negative fashion towards their noncleaved counterparts. Thus, caspaseaction not only reduces the amount of signalling intermediates necessary to transduce a NF-κB response, but also creates novel peptides that actively interfere with NF-κB activation. While IKK2, 247 p65, 250,251 I-κB 246, Akt 252 and HPK1 248 are mainly cleaved by caspase-3-related caspases during the effector stage of apoptosis, NIK, 249 RIP 243,244 and TRAF1 245 are cleaved by caspase-8 during the initiator phase of death receptor-induced apoptosis. Thus, caspase-8-generated cleavage products derived from RIP, TRAF1 and NIK may have a special role in TNF-R1-induced apoptosis by blocking the concomitantly induced anti-apoptotic NF-κB response. As death receptors are in principle able to activate both the apoptotic and the NF-κB pathway, the regulatory network of these pathways allows a highly flexible cellular behavior in response to stimulation of death receptors, especially TNF-R1. Aside from NF-κB activation, stimulation of c-Jun N-terminal kinase is a second cellular response to TNF in common to all cell types, which could play a role in apoptosisinduction by TNF-R1. As outlined in more detail in the following, there is evidence for an apoptosis-related crosstalk of the JNK pathway with both the NF-κB, and the apoptotic pathway itself.
Although c-Jun N-terminal kinase is robustly activated by TNF via TNF-R1 in almost every cell line investigated, the role of JNK for TNF function, especially TNF-mediated apoptosis, is still poorly understood. An essential role of the JNK signalling pathway in excitotoxic stress-induced neuronal apoptosis 253,254 and UV-stimulated apoptosis 255 has been clearly demonstrated, in particular in studies with mouse embryonic fibroblasts derived from JNK-deficient mice. The proapoptotic action of JNKs seem mainly dependent on their capability to phosphorylate c-Jun, a component of the heterodimeric transcription factor AP-1, 255 but can also be related to phosphorylation and inhibition of Bcl-2.256,257,258 Nevertheless, the JNK pathway can also have an anti-apoptotic function, for example during neuronal development 259 or in thymocytes.260 The ambivalent function of the JNK pathway in different apoptotic scenarios is also reflected in its role in TNF-induced apoptosis. While mouse embryonic fibroblasts of JNK1−/−-and JNK2−/−-deficient mice show increased sensitivity against TNF-induced apoptosis, 261 mouse embryonic fibroblasts of mice deficient for ASK1, an MAP3K implicated in TNF-R1-mediated JNK activation, are significantly protected against TNF-induced cell death.144 Moreover, studies in other cell types using inhibitors of the JNK pathway also revealed a proapoptotic function of the JNK pathway in TNF-induced cell death.262,263,264,265,266,267,268 The different roles of JNK in TNF-induced apoptosis could be partly related to celltype- specific effects, but may also mirror that TNF engages JNK by more than one pathway. Indeed, especially under apoptotic conditions, TNF activates JNK, but also p38, with biphasic kinetics.264 The first transient phase of JNK activation is caspase-independent 269,270 and almost completely inhibited in MKK7- deficient mouse embryonic fibroblasts.139 In contrast, the second phase of TNF-induced JNK activation correlates with apoptosis-induction and is blocked by caspase inhibitors. Remarkably, mouse embryonic fibroblasts of mice deficient of ASK1 show reduced sensitivity against TNF-induced apoptosis and a reduction in apoptosis-related delayed JNK activation, but are undisturbed with respect to transient rapid TNF-induced JNK.144 Thus, this delayed prolonged type of JNK activation by TNF may rather reflect a common response to the activation of the apoptotic pathway, as described elsewhere, 271,272 than being a genuine, direct TNF-mediated effect. Nevertheless, caspase-mediated activation of JNK could trigger a self-amplifying apoptotic loop, for example, by upregulation of death ligands. In agreement with this idea, a kinase-dead mutant of ASK1 weakens TNF-induced apoptosis.143,154,273 In addition, it has been recently shown that ASK1 can trigger a caspase-dependent, 274 but also a caspase-independent 275 pathway leading to cell death. In this regard, NF-κB activation inhibits prolonged TNF-induced JNK activation 267,268,276 and this has been attributed to the upregulation of JNK inhibitory proteins, XIAP1 267 in one study and GADD45β 276 in another study. Both molecules are identified and characterized under conditions, where TNF-induced NF-κB activation is compromised in the absence of caspase inhibitors. Therefore, it is possible that XIAP1 and GADD45ß are not directly involved in the regulation of JNK activity, but interfere with prolonged TNF-induced and ASK1-mediated JNK activation by blocking apoptosis and caspase activation as described above. A role of ASK1 in the delayed phase of TNF-induced JNK activation and apoptosis induction is also in agreement with the finding that ASK1 is activated by reactive oxygen species (ROS).156 Indeed, the generation of ROS can have a dual role in TNF signalling. On one hand, it can promote NF-κB activation, which is redox-sensitive 277 but on the other hand, ROS have also been implicated as mediators of TNF-induced apoptosis.278,279,280,281,282,283 Remarkably, manganous superoxide dismutase (MnSOD), which acts as a scavenger of potentially toxic superoxide radicals, is an NF-κB-dependent target gene of TNF.282,283 This emphasizes again that NF-κB activation and apoptosis signalling by TNF are tightly connected by a regulatory network (Figure 4). ROS-generating compounds can themselves induce caspase-activation and apoptosis via the mitochondrial pathway, in the context of TNF signalling ROS production seems to act as an amplification mechanism.
Noteworthy, production of ROS has not only a role in apoptosis but also in necrosis. This form of cell death is largely independent of caspases, morphologically quite different from apoptosis and in vivo associated with inflammation.284 Necrosis can be induced by death receptors including TNF-R1 by a RIP-dependent pathway.285 In contrast to its role in the NF-κB pathway, the role of RIP in induction of necron's is dependent on the kinase activity of the molecule.285 As FADD, but not caspase-8, is essentially involved in death receptor-induced necrosis, FADD seems to be the bifurcation point of death receptor-mediated apoptosis and necrosis.285 There is evidence that death receptor-induced apoptosis and necrosis do not simply act as parallel linear death pathways. Under apoptotic conditions caspase-mediated cleavage of RIP may block the necrotic pathway. Consequently, inhibition of the FADD-caspase axis of cell death by use of caspase inhibitors or FADD deletion mutants lacking their DED leads to sensitiation to death receptor-induced necrosis286,287,288,289. A pivotal role in mediating the necrotic response has been attributed to the production of ROS.288,290,291 How RIP is involved in the production of ROS and how this is related to the capability of TRAF2 to induce ROS155 is still an open question. TNF triggers oxidative stress (see also JNK) and mitochondria-dependent production of ROS.278,292,293,294 As a consequence of ROS production, DNA is damaged leading to the activation of PARP-1. Excessive action of PARP-1 consumes high amounts of NAD+ and leads to ATP depletion, creating thereby conditions that favor necrosis instead of apoptosis.295,296,297,298 Remarkably, PARP-1 is cleaved by caspases into an amino-terminal fragment retaining DNA-binding capabilities and a carboxy-terminal fragment without catalytic activity.299 In particular, the amino-terminal PARP-1 fragment acts as a dominant-negative variant for uncleaved PARP-1.299 As ATP is required for apoptosis, PARP-1 cleavage acts as a switch between apoptotic and necrotic cell death.300 The fact that Fas is a more robust activator of caspase-8 compared to TNF-R1 may explain why in some cells that undergo apoptosis in response to Fas stimulation, TNF-R1 triggers a necrotic form of cell death.300,301 Nevertheless, inhibition of the apoptotic pathway can unmask the capability of Fas to trigger the necrotic response.288,290
Several studies have implicated acidic SMase and ceramide release in TNF-induced cell death.171,302,303,304 As cells deficient for acidic SMase are still TNF sensitive,305 TNF-induced ceramide production seems to have no general, obligatory role in TNF-induced cell death. Nevertheless, the demonstration of reduced TNFsensitivity of aSMase−/− mouse embryonic fibroblasts306 and several other studies point to aSMase and ceramide production as celltype specific modulatory factors of TNF-induced cell death.307 TNF-R1-induced activation of aSMase occurs by a FADD-dependent, but caspase-8 independent pathway.308,309 In so far the pathway leading to aSMase activation is identical to the receptor-proximal necrotic pathway it remains to be verified to which extent the aSMAse pathway is related to the necrotic response. Ceramide release by aSMase can contribute to the activation of caspases and can, in addition, lead to the activation of the lysosomal compartment, in particular cathepsin D, which specifically binds ceramide.310 Although lysosomal enzymes have been implicated in necrosis (for review see 284) they can also activate caspases and induce apoptosis,311 most likely via Bid cleavage.312 The potential relevance of cathepsins for TNF-induced cell death313,314,315 is most obvious for cathepsin B, as mice deficient for this protease are resistant to TNF-mediated hepatocyte apoptosis and liver injury.316
Mechanisms of TNF-R2-induced apoptosis
Early studies with soluble TNF gave inconsistent results with respect to an apoptosis-inducing capability of the nondeath domain-containing receptor TNF-R2. 317,318,319 However, the use of agonistic TNF-R2-specific antibodies clearly showed that exclusive triggering of this receptor is in some cells sufficient to induce cell death.320,321,322 Analyses with TNF and TNF receptor-specific neutralizing antibodies showed that stimulation of TNF-R2 does not directly engage the apoptotic program, but relies on the induction of endogenous, membrane-bound TNF, which subsequently activates TNF-R1 (Figure 5).323,324 Remarkably, the action of the endogenously produced membrane-bound TNF on TNF-R1 is drastically enhanced by the existing stimulation of TNF-R2. Thus, apoptosis induction by the non-death domain-containing receptor TNF-R2 is not only dependent on the production of endogenous TNF and expression of TNF-R1, but also by the TNF-R2-dependent process described above leading to depletion of TRAF2 and TRAF2-associated protective factors. Similarly, apoptosis induction by the other members of the TNF receptor family lacking a death domain has also been attributed to the induction of endogenous TNF and concomitant depletion of TRAF2.323,324,325,326

Model of non-death domain-containing receptor-induced apoptosis. Non-death domain-containing receptors trigger two events that cooperatively induce apoptosis: (1) Inhibition of TRAF2-mediated NF-κB activation and deviation of TRAF2-containing antiapoptotic complexes (gray) and (ii) induction of endogenous membrane-TNF that stimulates in addition the death receptor TNF-R1
Abbreviations
- ASK1:
-
apoptosis signal-regulated kinase-1
- aSMase:
-
acidic sphingomyelinase
- BIR:
-
baculovirus IAP repeat
- DD:
-
death domain
- DED:
-
death effector domain
- DISC:
-
death-inducing signalling complex
- FADD:
-
Fas-associated death domain protein
- FAN:
-
factor associated with neutral sphingomyelinase activation
- GSK:
-
germinal center kinase
- IAP:
-
inhibitor of apoptosis protein
- IKK:
-
inhibitor of NF-κB kinase
- IL:
-
interleukin
- I-κB:
-
inhibitor of kappa B
- JNK:
-
c-Jun N-terminal kinase
- LPS:
-
lipopolysaccharide
- LTa:
-
lymphotoxin-a
- MAPK:
-
mitogen-activated protein kinase
- MEF:
-
mouse embryonic fibroblast
- MEKK:
-
mitogen-activated protein kinase/Erk kinase kinase
- NEMO:
-
NF-κB essential modulator
- NF-κB:
-
nuclear factor kappa B
- PARP:
-
poly(ADP-ribose) polymerase
- nSMase:
-
neutral sphingomyelinase
- PI3K:
-
phosphoinositide-3OH kinase
- PKA:
-
protein kinase A
- PKB:
-
protein kinase B
- PKC:
-
protein kinase C
- PLAD:
-
pre-ligand binding assembly domain
- PTEN:
-
phosphatase and tensin homologue deleted on chromosome 10
- RHD:
-
rel homology domain
- RING:
-
really interesting new gene
- RIP:
-
receptor-interacting protein
- ROS:
-
reactive oxygen species
- SAPK:
-
stress-activated protein kinase
- TACE:
-
TNF alpha-converting enzyme
- SODD:
-
silencer of death domain protein
- TANK:
-
TRAF-associated NF-kappaB activator
- TNF:
-
tumor necrosis factor
- TRADD:
-
TNF receptor-associated death domain protein
- TRAF:
-
TNF receptor-associated factor
- Trx:
-
thioredoxin
References
Kriegler M, Perez C, DeFay K et al. (1988) A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell 53: 45–53
Tang P, Hung M-C and Klostergaard J (1996) Human pro-tumor necrosis factor is a homotrimer. Biochemistry 35: 8216–8225
Black RA, Rauch CT, Kozlosky CJ et al. (1997) A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385: 729–733
Bazan JF (1993) Emerging families of cytokines and receptors. Curr. Biol. 3: 603–606
Locksley RM, Killeen N and Lenardo MJ (2001) The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104: 487–501
Naismith JH and Sprang SR (1998) Modularity in the TNF-receptor family. Trends Biochem. Sci. 23: 74–79
Banner DW, D'Arcy A, Janes W et al. (1993) Crystal structure of the soluble human 55 kd TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell 73: 431–445
Chan FK, Chun HJ, Zheng L et al. (2000) A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 288: 2351–2354
Grell M, Douni E, Wajant H et al. (1995) The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83: 793–802
Grell M, Wajant H, Zimmermann G and Scheurich P (1998) The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc. Natl. Acad. Sci. USA 95: 570–575
Wallach D, Engelmann H, Nophar Y et al. (1991) Soluble and cell surface receptors for tumor necrosis factor. Agents Actions Suppl. 35: 51–57
Taylor PC (2001) Anti-tumor necrosis factor therapies. Curr. Opin. Rheumatol. 13: 164–169
Solomon KA, Pesti N, Wu G and Newton RC (1999) Cutting edge: a dominant negative form of TNF-alpha converting enzyme inhibits proTNF and TNFRII secretion. J. Immunol. 163: 4105–4108
McDermott MF, Aksentijevich I, Galon J et al. (1999) Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 97: 133–144
Tartaglia LA, Ayres TM, Wong GH and Goeddel DV (1993) A novel domain within the 55 kd TNF receptor signals cell death. Cell 74: 845–853
Schulze-Osthoff K, Ferrari D, Los M et al. (1998) Apoptosis signaling by death receptors. Eur. J. Biochem. 254: 439–459
Mannel DN and Echtenacher B (2000) TNF in the inflammatory response. Chem. Immunol. 74: 141–161
Beutler B, Greenwald D, Hulmes JD et al. (1985) Identity of tumour necrosis factor and the macrophage-secreted factor cachectin. Nature 316: 552–554
Probert L, Akassoglou K, Pasparakis M et al. (1995) Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc. Natl. Acad. Sci. USA 92: 11294–11298
Fontaine V, Mohand-Said S, Hanoteau N et al. (2002) Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J. Neurosci. 22: RC216
Kontoyiannis D, Pasparakis M, Pizarro TT et al. (1999) Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 10: 387–398
Yamada Y, Kirillova I, Peschon JJ and Fausto N (1997) Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc. Natl. Acad. Sci. USA 94: 1441–1446
Bradham CA, Plumpe J, Manns MP et al. (1998) Mechanisms of hepatic toxicity. I. TNF-induced liver injury. Am. J. Physiol. 275: G387–G392
Taylor PC, Peters AM, Paleolog E et al. (2000) Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum. 43: 38–47
Blam ME, Stein RB and Lichtenstein GR (2001) Integrating anti-tumor necrosis factor therapy in inflammatory bowel disease: current and future perspectives. Am. J. Gastroenterol. 96: 1977–1997
Chaplin DD and Fu Y (1998) Cytokine regulation of secondary lymphoid organ development. Curr. Opin. Immunol. 10: 289–297
Ruddle NH (1999) Lymphoid neo-organogenesis: lymphotoxin's role in inflammation and development. Immunol. Res. 19: 119–125
Marino MW, Dunn A, Grail D et al. (1997) Characterization of tumor necrosis factor-deficient mice. Proc. Natl. Acad. Sci. USA 94: 8093–8098
Flynn JL, Goldstein MM, Chan J et al. (1995) Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2: 561–572
Rothe J, Mackay F, Bluethmann H et al. (1994) Phenotypic analysis of TNFR1-deficient mice and characterization of TNFR1-deficient fibroblasts in vitro. Circ. Shock 44: 51–56
Vieira LQ, Goldschmidt M, Nashleanas M et al. (1996) Mice lacking the TNF receptor p55 fail to resolve lesions caused by infection with Leishmania major, but control parasite replication. J. Immunol. 157: 827–835
Bean AG, Roach DR, Briscoe H et al. (1999) Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J. Immunol. 162: 3504–3511
Deckert-Schluter M, Bluethmann H, Rang A et al. (1998) Crucial role of TNF receptor type 1 (p55), but not of TNF receptor type 2 (p75), in murine toxoplasmosis. J. Immunol. 160: 3427–3436
Camelo S, Lafage M and Lafon M (2000) Absence of the p55 Kd TNF-alpha receptor promotes survival in rabies virus acute encephalitis. J. Neurovirol. 6: 507–518
Zhao YX, Lajoie G, Zhang H et al. (2000) Tumor necrosis factor receptor p55-deficient mice respond to acute Yersinia enterocolitica infection with less apoptosis and more effective host resistance. Infect. Immun. 68: 1243–1251
Lucas R, Juillard P, Decoster E et al. (1997) Crucial role of tumor necrosis factor (TNF) receptor 2 and membrane- bound TNF in experimental cerebral malaria. Eur. J. Immunol. 27: 1719–1725
Kollias G, Douni E, Kassiotis G and Kontoyiannis D (1999) On the role of tumor necrosis factor and receptors in models of multiorgan failure, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Immunol. Rev. 169: 175–194
Wagner TE, Huseby ES and Huseby JS (2002) Exacerbation of Mycobacterium tuberculosis enteritis masquerading as Crohn's disease after treatment with a tumor necrosis factor-alpha inhibitor. Am. J. Med. 112: 67–69
Old LJ (1988) Tumor necrosis factor. Sci. Am. 258: 59–75
Sugarman BJ, Aggarwal BB, Hass PE et al. (1985) Recombinant human tumor necrosis factor-alpha: effects on proliferation of normal and transformed cells in vitro. Science 230: 943–945
Hock H, Dorsch M, Kunzendorf U et al. (1993) Mechanisms of rejection induced by tumor cell-targeted gene transfer of interleukin 2, interleukin 4, interleukin 7, tumor necrosis factor, or interferon gamma. Proc. Natl. Acad. Sci. USA 90: 2774–2778
Eggermont AM and ten Hagen TL (2001) Isolated limb perfusion for extremity soft-tissue sarcomas, in-transit metastases, and other unresectable tumors: credits, debits, and future perspectives. Curr. Oncol. Rep. 3: 359–367
Ruegg C, Yilmaz A and Bieler G et al. (1998) Evidence for the involvement of endothelial cell integrin alphaVbeta3 in the disruption of the tumor vasculature induced by TNF and IFN-gamma. Nat. Med. 4: 408–414
Baud V and Karin M (2001) Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 11: 372–377
Verma IM, Stevenson JK, Schwarz EM et al. (1995) Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 9: 2723–2735
Perkins ND (2000) The Rel/NF-kappa B family: friend and foe. Trends Biochem. Sci. 25: 434–440
Mercurio F, Zhu H, Murray BW et al. (1997) IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF- kappaB activation. Science 278: 860–866
DiDonato JA, Hayakawa M, Rothwarf DM et al. (1997) A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 388: 548–554
Zandi E, Rothwarf DM, Delhase M et al. (1997) The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 91: 243–252
Yamaoka S, Courtois G, Bessia C et al. (1998) Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell 93: 1231–1240
Li Y, Kang J, Friedman J et al. (1999) Identification of a cell protein (FIP-3) as a modulator of NF-kappaB activity and as a target of an adenovirus inhibitor of tumor necrosis factor alpha-induced apoptosis. Proc. Natl. Acad. Sci. USA 96: 1042–1047
Rothwarf DM, Zandi E, Natoli G and Karin M (1998) IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 395: 297–300
Harhaj EW, Good L, Xiao G et al. (2000) Somatic mutagenesis studies of NF-kappa B signaling in human T cells: evidence for an essential role of IKK gamma in NF-kappa B activation by T-cell costimulatory signals and HTLV-I Tax protein. Oncogene 19: 1448–1456
Mercurio F, Murray BW, Shevchenko A et al. (1999) IkappaB kinase (IKK)-associated protein 1, a common component of the heterogeneous IKK complex. Mol. Cell Biol. 19: 1526–1538
Chen G, Cao P and Goeddel DV (2002) TNF-induced recruitment and activation of the IKK complex require Cdc37 and Hsp90. Mol. Cell 9: 401–410
Rudolph D, Yeh WC, Wakeham A et al. (2000) Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev. 14: 854–862
Schmidt-Supprian M, Bloch W, Courtois G et al. (2000) NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol. Cell 5: 981–992
Makris C, Godfrey VL, Krahn-Senftleben G et al. (2000) Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol. Cell 5: 969–979
Tanaka M, Fuentes ME, Yamaguchi K et al. (1999) Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity 10: 421–429
Li Q, Van Antwerp D, Mercurio F et al. (1999) Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science 284: 321–325
Takeda K, Takeuchi O, Tsujimura T et al. (1999) Limb and skin abnormalities in mice lacking IKKalpha. Science 284: 313–316
Hu Y, Baud V, Delhase M et al. (1999) Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science 284: 316–320
Li Q, Lu Q, Hwang JY et al. (1999) IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 13: 1322–1328
Senftleben U, Cao Y, Xiao G et al. (2001) Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 293: 1495–1499
Xiao G, Cvijic ME, Fong A et al. (2001) Retroviral oncoprotein Tax induces processing of NF-kappaB2/p100 in T cells: evidence for the involvement of IKKalpha. EMBO J. 20: 6805–6815
Li Q, Estepa G, Memet S et al. (2000) Complete lack of NF-kappaB activity in IKK1 and IKK2 double-deficient mice: additional defect in neurulation. Genes Dev. 14: 1729–1733
Pomerantz JL and Baltimore D (1999) NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 18: 6694–6704
Bonnard M, Mirtsos C, Suzuki S et al. (2000) Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 19: 4976–4985
Tojima Y, Fujimoto A, Delhase M et al. (2000) NAK is an IkappaB kinase-activating kinase. Nature 404: 778–782
Peters RT, Liao SM and Maniatis T (2000) IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol. Cell 5: 513–522
Jiang Y, Woronicz JD, Liu W and Goeddel DV (1999) Prevention of constitutive TNF receptor 1 signaling by silencer of death domains. Science 283: 543–546
Hsu H, Xiong J and Goeddel DV (1995) The TNF receptor 1-associated protein TRADD signals cell death and NF- kappa B activation. Cell 81: 495–504
Hsu H, Huang J, Shu HB et al. (1996) TNF-dependent recruitment of the protein kinase RIP to the TNF receptor- 1 signaling complex. Immunity 4: 387–396
Wajant H, Henkler F, Scheurich P (2001) The TNF-receptor-associated factor family. Scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal. 13: 389–400
Hsu H, Shu HB, Pan MG and Goeddel DV (1996) TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84: 299–308
Devin A, Cook A, Lin Y et al. (2000) The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity 12: 419–429
Zhang SQ, Kovalenko A, Cantarella G and Wallach D (2000) Recruitment of the IKK signalosome to the p55 TNF receptor: RIP and A20 bind to NEMO (IKKgamma) upon receptor stimulation. Immunity 12: 301–311
Devin A, Lin Y, Yamaoka S et al. (2001) The alpha and beta subunits of IkappaB kinase (IKK) mediate TRAF2- dependent IKK recruitment to tumor necrosis factor (TNF) receptor 1 in response to TNF. Mol. Cell Biol. 21: 3986–3994
Ting AT, Pimentel-Muinos FX and Seed B (1996) RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 15: 6189–6196
Yang J, Lin Y, Guo Z et al. (2001) The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat. Immunol. 2: 620–624
Baud V, Liu ZG, Bennett B et al. (1999) Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 13: 1297–1308
Kim JW, Joe CO and Choi EJ (2001) Role of receptor-interacting protein in tumor necrosis factor-alpha-dependent MEKK1 activation. J. Biol. Chem. 276: 27064–27070
Xia Y, Makris C, Su B et al. (2000) MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc. Natl. Acad. Sci. USA 97: 5243–5248
Yujiri T, Ware M, Widmann C et al. (2000) MEK kinase 1 gene disruption alters cell migration and c-Jun NH2- terminal kinase regulation but does not cause a measurable defect in NF- kappa B activation. Proc. Natl. Acad. Sci. USA 97: 7272–7277
Sanz L, Diaz-Meco MT, Nakano H and Moscat J (2000) The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL1-TRAF6 pathway. EMBO J. 19: 1576–1586
Leitges M, Sanz L, Martin P et al. (2001) Targeted disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway. Mol. Cell 8: 771–780
Anrather J, Csizmadia V, Soares MP and Winkler H (1999) Regulation of NF-kappaB RelA phosphorylation and transcriptional activity by p21(ras) and protein kinase Czeta in primary endothelial cells. J. Biol. Chem. 274: 13594–13603
Zhong H, SuYang H, Erdjument-Bromage H et al. (1997) The transcriptional activity of NF-kappaB is regulated by the IkappaB- associated PKAc subunit through a cyclic AMP-independent mechanism. Cell 89: 413–424
Lallena MJ, Diaz-Meco MT, Bren G et al. (1999) Activation of IkappaB kinase beta by protein kinase C isoforms. Mol. Cell Biol. 19: 2180–2188
Sanz L, Sanchez P, Lallena MJ et al. (1999) The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 18: 3044–3053
Wang D, Westerheide SD, Hanson JL and Baldwin AS (2000) Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J. Biol. Chem. 275: 32592–32597
Sizemore N, Lerner N, Dombrowski N et al. (2002) Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B. J. Biol. Chem. 277: 3863–3869
Ozes ON, Mayo LD, Gustin JA et al. (1999) NF-kappaB activation by tumour necrosis factor requires the Akt serine–threonine kinase. Nature 401: 82–85
Romashkova JA and Makarov SS (1999) NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 401: 86–90
Hanna AN, Chan EY, Xu J et al. (1999) A novel pathway for tumor necrosis factor-alpha and ceramide signaling involving sequential activation of tyrosine kinase, p21(ras), and phosphatidylinositol 3-kinase. J. Biol. Chem. 274: 12722–12729
Kim BC, Lee MN, Kim JY et al. (1999) Roles of phosphatidylinositol 3-kinase and Rac in the nuclear signaling by tumor necrosis factor-alpha in rat-2 fibroblasts. J. Biol. Chem. 274: 24372–24377
Reddy SA, Huang JH and Liao WS (2000) Phosphatidylinositol 3-kinase as a mediator of TNF-induced NF-kappa B activation. J. Immunol. 164: 1355–1363
Pastorino JG, Tafani M and Farber JL (1999) Tumor necrosis factor induces phosphorylation and translocation of BAD through a phosphatidylinositide-3-OH kinase-dependent pathway. J. Biol. Chem. 274: 19411–19416
Plo I, Lautier D, Levade T et al. (2000) Phosphatidylcholine-specific phospholipase C and phospholipase D are respectively implicated in mitogen-activated protein kinase and nuclear factor kappaB activation in tumour-necrosis-factor-alpha-treated immature acute-myeloid-leukaemia cells. Biochem. J. 351: 459–467
Osawa Y, Banno Y, Nagaki M et al. (2001) TNF-alpha-induced sphingosine 1-phosphate inhibits apoptosis through a phosphatidylinositol 3-kinase/Akt pathway in human hepatocytes. J. Immunol. 167: 173–180
Martin AG, San Antonio B and Fresno M (2001) Regulation of nuclear factor kappa B transactivation. Implication of phosphatidylinositol 3-kinase and protein kinase C zeta in c-Rel activation by tumor necrosis factor alpha. J. Biol. Chem. 276: 15840–15849
Mayo MW, Madrid LV, Westerheide SD et al. (2002) PTEN blocks tumor necrosis factor-induced NF-kappa B-dependent transcription by inhibiting the transactivation potential of the p65 subunit. J. Biol. Chem. 277: 11116–11125
Gustin JA, Maehama T, Dixon JE and Donner DB . (2001) The PTEN tumor suppressor protein inhibits tumor necrosis factor- induced nuclear factor kappa B activity. J. Biol. Chem. 276: 27740–27744
Madrid LV, Mayo MW, Reuther JY and Baldwin Jr AS (2001) Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J. Biol. Chem. 276: 18934–18940
Burkle A (2001) Poly(APD-ribosyl)ation, a DNA damage-driven protein modification and regulator of genomic instability. Cancer Lett. 163: 1–5
Oliver FJ, Menissier-de Murcia J, Nacci C et al. (1999) Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 18: 4446–4454
Hassa PO and Hottiger MO (1999) A role of poly (ADP-ribose) polymerase in NF-kappaB transcriptional activation. Biol. Chem. 380: 953–959
Hassa PO, Covic M, Hasan S et al. (2001) The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J. Biol. Chem. 276: 45588–45597
Chang WJ and Alvarez-Gonzalez R (2001) The sequence-specific DNA binding of NF-kappa B is reversibly regulated by the automodification reaction of poly (ADP-ribose) polymerase 1. J. Biol. Chem. 276: 47664–47670
Ullrich O, Diestel A, Eyupoglu IY, Nitsch R (2001) Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat. Cell Biol. 3: 1035–1042
Le Page C, Sanceau J, Drapier JC, Wietzerbin J (1998) Inhibitors of ADP-ribosylation impair inducible nitric oxide synthase gene transcription through inhibition of NF kappa B activation. Biochem. Biophys. Res. Commun. 243: 451–457
Kameoka M, Ota K, Tetsuka T et al. (2000) Evidence for regulation of NF-kappaB by poly(ADP-ribose) polymerase. Biochem. J. 346: 641–649
Ha HC, Hester LD, Snyder SH (2002) Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc. Natl. Acad. Sci. USA 99: 3270–3275
Hoeflich KP, Luo J, Rubie EA et al. (2000) Requirement for glycogen synthase kinase-3beta in cell survival and NF- kappaB activation. Nature 406: 86–90
Higuchi M and Aggarwal BB (1994) TNF induces internalization of the p60 receptor and shedding of the p80 receptor. J. Immunol. 152: 3550–3558
Mosselmans R, Hepburn A, Dumont JE et al. (1988) Endocytic pathway of recombinant murine tumor necrosis factor in L-929 cells. J. Immunol. 141: 3096–3100
Schutze S, Machleidt T, Adam D et al. (1999) Inhibition of receptor internalization by monodansylcadaverine selectively blocks p55 tumor necrosis factor receptor death domain signaling. J. Biol. Chem. 274: 10203–10212
Beg AA, Sha WC, Bronson RT and Baltimore D (1995) Constitutive NF-kappa B activation, enhanced granulopoiesis, and neonatal lethality in I kappa B alpha-deficient mice. Genes Dev. 9: 2736–2746
Sarma V, Lin Z, Clark L et al. (1995) Activation of the B-cell surface receptor CD40 induces A20, a novel zinc finger protein that inhibits apoptosis. J. Biol. Chem. 270: 12343–12346
Song HY, Rothe M and Goeddel DV (1996) The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proc. Natl. Acad. Sci. USA 93: 6721–6725
Lee EG, Boone DL, Chai S et al. (2000) Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 289: 2350–2354
Zetoune FS, Murthy AR, Shao Z et al. (2001) A20 inhibits NF-kappa B activation downstream of multiple Map3 kinases and interacts with the I kappa B signalosome. Cytokine 15: 282–298
Heyninck K, De Valck D, Vanden Berghe W et al. (1999) The zinc finger protein A20 inhibits TNF-induced NF-kappaB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-kappaB- inhibiting protein ABIN. J. Cell Biol. 145: 1471–1482
Jaattela M, Mouritzen H, Elling F and Bastholm L (1996) A20 zinc finger protein inhibits TNF and IL1 signaling. J. Immunol. 156: 1166–1173
Delhase M, Hayakawa M, Chen Y and Karin M (1999) Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science 284: 309–313
Shaulian E and Karin M (2002) AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4: 131–136
Chang L and Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410: 37–40
Brenner DA, O'Hara M, Angel P et al. (1989) Prolonged activation of jun and collagenase genes by tumour necrosis factor-alpha. Nature 337: 661–663
Hanazawa S, Takeshita A, Amano S et al. (1993) Tumor necrosis factor-alpha induces expression of monocyte chemoattractant JE via fos and jun genes in clonal osteoblastic MC3T3- E1 cells. J. Biol. Chem. 268: 9526–9532
Min W and Pober JS (1997) TNF initiates E-selectin transcription in human endothelial cells through parallel TRAF-NF-kappa B and TRAF-RAC/CDC42-JNK-c-Jun/ATF2 pathways. J. Immunol. 159: 3508–3518
Diehl AM, Yin M, Fleckenstein J et al. (1994) Tumor necrosis factor-alpha induces c-jun during the regenerative response to liver injury. Am. J. Physiol. 267: G552–G561
Bruccoleri A, Gallucci R, Germolec DR et al. (1997) Induction of early-immediate genes by tumor necrosis factor alpha contribute to liver repair following chemical-induced hepatotoxicity. Hepatology 25: 133–141
Reinhard C, Shamoon B, Shyamala V and Williams LT (1997) Tumor necrosis factor alpha-induced activation of c-jun N-terminal kinase is mediated by TRAF2. EMBO J. 16: 1080–1092
Natoli G, Costanzo A, Ianni A et al. (1997) Activation of SAPK/JNK by TNF receptor 1 through a noncytotoxic TRAF2- dependent pathway. Science 275: 200–203
Liu ZG, Hsu H, Goeddel DV, Karin M (1996) Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 87: 565–576
Song HY, Regnier CH, Kirschning CJ et al. (1997) Tumor necrosis factor (TNF)-mediated kinase cascades: bifurcation of nuclear factor-kappaB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proc. Natl. Acad. Sci. USA 94: 9792–9796
Lee SY, Reichlin A, Santana A et al. (1997) TRAF2 is essential for JNK but not NF-kappaB activation and regulates lymphocyte proliferation and survival. Immunity 7: 703–713
Yeh WC, Shahinian A, Speiser D et al. (1997) Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity 7: 715–725
Tournier C, Dong C, Turner TK et al. (2001) MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 15: 1419–1426
Lawler S, Fleming Y, Goedert M and Cohen P (1998) Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro. Curr. Biol. 8: 1387–1390
Moriguchi T, Toyoshima F, Masuyama N et al. (1997) A novel SAPK/JNK kinase, MKK7, stimulated by TNFalpha and cellular stresses. EMBO J. 16: 7045–7053
Nishitoh H, Saitoh M, Mochida Y et al. (1998) ASK1 is essential for JNK/SAPK activation by TRAF2. Mol. Cell 2: 389–395
Hoeflich KP, Yeh WC, Yao Z et al. (1999) Mediation of TNF receptor-associated factor effector functions by apoptosis signal-regulating kinase-1 (ASK1). Oncogene 18: 5814–5820
Tobiume K, Matsuzawa A, Takahashi T et al. (2001) ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2: 222–228
Kyriakis JM (1999) Signaling by the germinal center kinase family of protein kinases. J. Biol. Chem. 274: 5259–5262
Yuasa T, Ohno S, Kehrl JH and Kyriakis JM (1998) Tumor necrosis factor signaling to stress-activated protein kinase (SAPK)/Jun NH2-terminal kinase (JNK) and p38. Germinal center kinase couples TRAF2 to mitogen-activated protein kinase/ERK kinase kinase 1 and SAPK while receptor interacting protein associates with a mitogen- activated protein kinase kinase kinase upstream of MKK6 and p38. J. Biol. Chem. 273: 22681–22692
Diener K, Wang XS, Chen C et al. (1997) Activation of the c-Jun N-terminal kinase pathway by a novel protein kinase related to human germinal center kinase. Proc. Natl. Acad. Sci. USA 94: 9687–9692
Shi CS and Kehrl JH (1997) Activation of stress-activated protein kinase/cJun N-terminal kinase, but not NF-kappaB, by the tumor necrosis factor (TNF) receptor 1 through a TNF receptor-associated factor 2- and germinal center kinase related-dependent pathway. J. Biol. Chem. 272: 32102–32107
Yao Z, Zhou G, Wang XS et al. (1999) A novel human STE20-related protein kinase, HGK, that specifically activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 274: 2118–2125
Nakano K, Yamauchi J, Nakagawa K et al. (2000) NESK, a member of the germinal center kinase family that activates the c-Jun N-terminal kinase pathway and is expressed during the late stages of embryogenesis. J. Biol. Chem. 275: 20553–20559
Fu CA, Shen M, Huang BC et al. (1999) TNIK, a novel member of the germinal center kinase family that activates the c-Jun N-terminal kinase pathway and regulates the cytoskeleton. J. Biol. Chem. 274: 30729–30737
Shi CS, Leonardi A, Kyriakis J et al. (1999) TNF-mediated activation of the stress-activated protein kinase pathway: TNF receptor-associated factor 2 recruits and activates germinal center kinase related. J. Immunol. 163: 3279–3285
Chadee DN, Yuasa T and Kyriakis JM (2002) Direct activation of mitogen-activated protein kinase kinase kinase MEKK1 by the Ste20p homologue GCK and the adapter protein TRAF2. Mol. Cell Biol. 22: 737–749
Ichijo H, Nishida E, Irie K et al. (1997) Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275: 90–94
Chandel NS, Schumacker PT and Arch RH (2001) Reactive oxygen species are downstream products of TRAF-mediated signal transduction. J. Biol. Chem. 276: 42728–42736
Gotoh Y and Cooper JA (1998) Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-alpha signal transduction. J. Biol. Chem. 273: 17477–17482
Saitoh M, Nishitoh H, Fujii M et al. (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 17: 2596–2606
Matsuda M, Masutani H, Nakamura H et al. (1991) Protective activity of adult T cell leukemia-derived factor (ADF) against tumor necrosis factor-dependent cytotoxicity on U937 cells. J. Immunol. 147: 3837–3841
Liu H, Nishitoh H, Ichijo H and Kyriakis JM (2000) Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol. Cell Biol. 20: 2198–2208
Carpentier I, Declercq W, Malinin NL et al. (1998) TRAF2 plays a dual role in NF-kappaB-dependent gene activation by mediating the TNF-induced activation of p38 MAPK and IkappaB kinase pathways. FEBS Lett. 425: 195–198
Pombo CM, Kehrl JH, Sanchez I et al. (1995) Activation of the SAPK pathway by the human STE20 homologue germinal centre kinase. Nature 377: 750–754
Kelliher MA, Grimm S, Ishida Y et al. (1998) The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8: 297–303
Wysk M, Yang DD, Lu HT et al. (1999) Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for tumor necrosis factor-induced cytokine expression. Proc. Natl. Acad. Sci. USA 96: 3763–3768
Martin-Blanco E (2000) p38 MAPK signalling cascades: ancient roles and new functions. Bioessays 22: 637–645
Winzen R, Kracht M, Ritter B et al. (1999) The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 18: 4969–4980
Miyazawa K, Mori A, Miyata H et al. (1998) Regulation of interleukin1beta-induced interleukin-6 gene expression in human fibroblast-like synoviocytes by p38 mitogen-activated protein kinase. J. Biol. Chem. 273: 24832–24838
Vanden Berghe W, Plaisance S, Boone E et al. (1998) p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways are required for nuclear factor-kappaB p65 transactivation mediated by tumor necrosis factor. J. Biol. Chem. 273: 3285–3290
Beyaert R, Cuenda A, Vanden Berghe W et al. (1996) The p38/RK mitogen-activated protein kinase pathway regulates interleukin-6 synthesis response to tumor necrosis factor. EMBO J. 15: 1914–1923
Adam-Klages S, Adam D, Wiegmann K et al. (1996) FAN, a novel WD-repeat protein, couples the p55 TNF-receptor to neutral sphingomyelinase. Cell 86: 937–947
Kreder D, Krut O, Adam-Klages S et al. (1999) Impaired neutral sphingomyelinase activation and cutaneous barrier repair in FAN-deficient mice. EMBO J. 18: 2472–2479
Bourteele S, Hausser A, Doppler H et al. (1998) Tumor necrosis factor induces ceramide oscillations and negatively controls sphingolipid synthases by caspases in apoptotic Kym-1 cells. J. Biol. Chem. 273: 31245–31251
Segui B, Cuvillier O, Adam-Klages S et al. (2001) Involvement of FAN in TNF-induced apoptosis. J. Clin. Invest. 108: 143–151
Luschen S, Adam D, Ussat S et al. (2000) Activation of ERK1/2 and cPLA(2) by the p55 TNF receptor occurs independently of FAN. Biochem. Biophys. Res. Commun. 274: 506–512
Boldin MP, Mett IL, Wallach D (1995) A protein related to a proteasomal subunit binds to the intracellular domain of the p55 TNF receptor upstream to its ‘death domain’. FEBS Lett. 367: 39–44
Tsurumi C, Shimizu Y, Saeki M et al. (1996) cDNA cloning and functional analysis of the p97 subunit of the 26S proteasome, a polypeptide identical to the type-1 tumor-necrosis-factor-receptor-associated protein-2/55.11. Eur. J. Biochem. 239: 912–921
Dunbar JD, Song HY, Guo D et al. (1997) Two-hybrid cloning of a gene encoding TNF receptor-associated protein 2, a protein that interacts with the intracellular domain of the type 1 TNF receptor: identity with subunit 2 of the 26S protease. J. Immunol. 158: 4252–4259
Song HY, Dunbar JD, Zhang YX et al. (1995) Identification of a protein with homology to hsp90 that binds the type 1 tumor necrosis factor receptor. J. Biol. Chem. 270: 3574–3581
Felts SJ, Owen BA, Nguyen P et al. (2000) The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J. Biol. Chem. 275: 3305–3312
Van Lint J, Agostinis P, Vandevoorde V et al. (1992) Tumor necrosis factor stimulates multiple serine/threonine protein kinases in Swiss 3T3 and L929 cells. Implication of casein kinase-2 and extracellular signal-regulated kinases in the tumor necrosis factor signal transduction pathway. J. Biol. Chem. 267: 25916–25921
Vietor I, Schwenger P, Li W et al. (1993) Tumor necrosis factor-induced activation and increased tyrosine phosphorylation of mitogenactivated protein (MAP) kinase in human fibroblasts. J. Biol. Chem. 268: 18994–18999
Muller G, Storz P, Bourteele S et al. (1998) Regulation of Raf-1 kinase by TNF via its second messenger ceramide and cross-talk with mitogenic signalling. EMBO J. 17: 732–742
Jupp OJ, McFarlane SM, Anderson HM et al. (2001) Type II tumour necrosis factor-alpha receptor (TNFR2) activates c-Jun N- terminal kinase (JNK) but not mitogen-activated protein kinase (MAPK) or p38 MAPK pathways. Biochem. J. 359: 525–535
Schievella AR, Chen JH, Graham JR and Lin LL (1997) MADD, a novel death domain protein that interacts with the type 1 tumor necrosis factor receptor and activates mitogen-activated protein kinase. J. Biol. Chem. 272: 12069–12075
Brown TL and Howe PH (1998) MADD is highly homologous to a Rab3 guanine-nucleotide exchange protein (Rab3-GEP). Curr. Biol. 8: R191
Chow VT and Lee SS (1996) DENN, a novel human gene differentially expressed in normal and neoplastic cells. DNA Seq. 6: 263–273
Hildt E and Oess S (1999) Identification of Grb2 as a novel binding partner of tumor necrosis factor (TNF) receptor I. J. Exp. Med., 189: 1707–1714
Yao B, Zhang Y, Delikat S et al. (1995) Phosphorylation of Raf by ceramide-activated protein kinase. Nature 378: 307–310
Lee SE, Chung WJ, Kwak HB et al. (2001) Tumor necrosis factor-alpha supports the survival of osteoclasts through the activation of Akt and ERK. J. Biol. Chem. 276: 49343–49349
Landry DW and Oliver JA (2001) The pathogenesis of vasodilatory shock. N. Engl. J. Med. 345: 588–595
Wajant H, Haas E, Schwenzer R et al. (2000) Inhibition of death receptor-mediated gene induction by a cycloheximide- sensitive factor occurs at the level of or upstream of Fas-associated death domain protein (FADD). J. Biol. Chem. 275: 24357–24366
Varfolomeev EE, Schuchmann M, Luria V et al. (1998) Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9: 267–276
Sanchez I, Xu CJ, Juo P et al. (1999) Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron 22: 623–633
Zhang J, Cado D, Chen A et al. (1998) Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature 392: 296–300
Yeh WC, Pompa JL, McCurrach ME et al. (1998) FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279: 1954–1958
Kischkel FC, Lawrence DA, Chuntharapai A et al. (2000) Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 12: 611–620
Sprick MR, Weigand MA, Rieser E et al. (2000) FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2 1669. Immunity 12: 599–609
Bodmer JL, Holler N, Reynard S et al. (2000) TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat. Cell Biol. 2: 241–243
Juo P, Woo MS, Kuo CJ et al. (1999) FADD is required for multiple signaling events downstream of the receptor Fas. Cell Growth Differ. 10: 797–804
Juo P, Kuo CJ, Yuan J and Blenis J (1998) Essential requirement for caspase-8/FLICE in the initiation of the Fas- induced apoptotic cascade. Curr. Biol. 8: 1001–1008
Chinnaiyan AM, O'Rourke K, Tewari M, and Dixit VM (1995) FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 81: 505–512
Boldin MP, Goncharov TM, Goltsev YV and Wallach D (1996) Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 85: 803–815
Muzio M, Chinnaiyan AM, Kischkel FC et al. (1996) FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--inducing signaling complex. Cell 85: 817–827
Algeciras-Schimnich A, Shen L, Barnhart BC et al. (2002) Molecular ordering of the initial signaling events of CD95. Mol. Cell Biol. 22: 207–220
Weiss T, Grell M, Hessabi B et al. (1997) Enhancement of TNF receptor p60-mediated cytotoxicity by TNF receptor p80: requirement of the TNF receptor-associated factor-2 binding site. J. Immunol. 158: 2398–2404
Weiss T, Grell M, Siemienski K et al. (1998) TNFR80-dependent enhancement of TNFR60-induced cell death is mediated by TNFR-associated factor 2 and is specific for TNFR60. J. Immunol. 161: 3136–3142
Duckett CS and Thompson CB (1997) CD30-dependent degradation of TRAF2: implications for negative regulation of TRAF signaling and the control of cell survival. Genes Dev. 11: 2810–2821
Li X, Yang Y and Ashwell JD (2002) TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature 416: 345–347
Chan FK and Lenardo MJ (2000) A crucial role for p80 TNF-R2 in ampli-fying p60 TNF-R1 apoptosis signals in T lymphocytes. Eur. J. Immunol. 30: 652–660
Brown KD, Hostager BS and Bishop GA (2002) Regulation of TRAF2 signaling by self-induced degradation. J. Biol. Chem. 277: 19433–19438
Fotin-Mleczek M, Henkler F, Samel D et al. (2002) Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J. Cell Sci. 115: 2757–2770
Shu HB, Takeuchi M and Goeddel DV (1996) The tumor necrosis factor receptor 2 signal transducers TRAF2 and c- IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc. Natl. Acad. Sci. USA 93: 13973–13978
Rothe M, Pan MG, Henzel WJ et al. (1995) The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 83: 1243–1252
Clem RJ (2001) Baculoviruses and apoptosis: the good, the bad, and the ugly. Cell Death Differ. 8: 137–143
Roy N, Deveraux QL, Takahashi R et al. (1997) The c-IAP-1 and c-IAP-2 proteins are direct inhibitors of specific caspases. EMBO J. 16: 6914–6925
Wang CY, Mayo MW, Korneluk RG et al. (1998) NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c- IAP2 to suppress caspase-8 activation. Science 281: 1680–1683
Yang Y, Fang S, Jensen JP et al. (2000) Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 288: 874–877
Huang H, Joazeiro CA, Bonfoco E et al. (2000) The inhibitor of apoptosis, cIAP2, functions as a ubiquitin–protein ligase and promotes in vitro ubiquitination of caspases-3 and -7. J. Biol. Chem. 275: 26661–26664
Luo X, Budihardjo I, Zou H et al. (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94: 481–490
Li H, Zhu H, Xu CJ and Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94: 491–501
Chai J, Du C, Wu JW et al. (2000) Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 406: 855–862
Verhagen AM, Ekert PG, Pakusch M et al. (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102: 43–53
Wei MC, Zong WX, Cheng EH et al. (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730
Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev. 15: 2922–2933
Tang D, Lahti JM and Kidd VJ (2000) Caspase-8 activation and bid cleavage contribute to MCF7 cellular execution in a caspase-3-dependent manner during staurosporine-mediated apoptosis. J. Biol. Chem. 275: 9303–9307
Engels IH, Stepczynska A, Stroh C et al. (2000) Caspase-8/FLICE functions as an executioner caspase in anticancer drug-induced apoptosis. Oncogene 19: 4563–4573
Wieder T, Essmann F, Prokop A et al. (2001) Activation of caspase-8 in drug-induced apoptosis of B-lymphoid cells is independent of CD95/Fas receptor–ligand interaction and occurs downstream of caspase-3. Blood 97: 1378–1387
Scaffidi C, Fulda S, Srinivasan A et al. (1998) Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 17: 1675–1687
Schwenzer R, Siemienski K, Liptay S et al. (1999) The human tumor necrosis factor (TNF) receptor-associated factor 1 gene (TRAF1) is up-regulated by cytokines of the TNF ligand family and modulates TNF-induced activation of NF-kappaB and c-Jun N-terminal kinase. J. Biol. Chem. 274: 19368–19374
Chu ZL, McKinsey TA, Liu L et al. (1997) Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc. Natl. Acad. Sci. USA 94: 10057–10062
Kreuz S, Siegmund D, Scheurich P and Wajant H (2001) NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell Biol. 21: 3964–3973
Micheau O, Lens S, Gaide O et al. (2001) NF-kappaB signals induce the expression of c-FLIP. Mol. Cell Biol. 21: 5299–5305
Wu MX, Ao Z, Prasad KV et al. (1998) IEX-1L, an apoptosis inhibitor involved in NF-kappaB-mediated cell survival. Science 281: 998–1001
Wang CY, Guttridge DC, Mayo MW and Baldwin Jr AS (1999) NF-kappaB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol. Cell Biol. 19: 5923–5929
Lee HH, Dadgostar H, Cheng Q et al. (1999) NF-kappaB-mediated up-regulation of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc. Natl. Acad. Sci. USA 96: 9136–9141
Zong WX, Edelstein LC, Chen C et al. (1999) The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kappaB that blocks TNFalpha-induced apoptosis. Genes Dev. 13: 382–387
Stehlik C, de Martin R, Kumabashiri I et al. (1998) Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha- induced apoptosis. J. Exp. Med. 188: 211–216
Tamatani M, Che YH, Matsuzaki H et al. (1999) Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J. Biol. Chem. 274: 8531–8538
Chen C, Edelstein LC and Gelinas C (2000) The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L). Mol. Cell Biol. 20: 2687–2695
Bernard D, Quatannens B, Vandenbunder B and Abbadie C (2001) Rel/NF-kappa B transcription factors protect against tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced apoptosis by up-regulating the TRAIL decoy receptor DcR1. J. Biol. Chem. 276: 27322–27328
Brunet A, Bonni A, Zigmond MJ et al. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868
Cardone MH, Roy N, Stennicke HR et al. (1998) Regulation of cell death protease caspase-9 by phosphorylation. Science 282: 1318–1321
del Peso L, Gonzalez-Garcia M, Page C et al. (1997) Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278: 687–689
Lin Y, Devin A, Rodriguez Y, Liu ZG (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF- induced apoptosis. Genes Dev. 13: 2514–2526
Martinon F, Holler N, Richard C and Tschopp J (2000) Activation of a pro-apoptotic amplification loop through inhibition of NF-kappaB-dependent survival signals by caspase-mediated inactivation of RIP. FEBS Lett. 468: 134–136
Irmler M, Steiner V, Ruegg C et al. (2000) Caspase-induced inactivation of the anti-apoptotic TRAF1 during Fas ligand-mediated apoptosis. FEBS Lett. 468: 129–133
Barkett M, Xue D, Horvitz HR and Gilmore TD (1997) Phosphorylation of IkappaB-alpha inhibits its cleavage by caspase CPP32 in vitro. J. Biol. Chem. 272: 29419–29422
Tang G, Yang J, Minemoto Y and Lin A (2001) Blocking caspase-3-mediated proteolysis of IKKbeta suppresses TNF-alpha- induced apoptosis. Mol. Cell 8: 1005–1016
Arnold R, Liou J, Drexler HC et al. (2001) Caspase-mediated cleavage of hematopoietic progenitor kinase 1 (HPK1) converts an activator of NFkappaB into an inhibitor of NFkappaB. J. Biol. Chem. 276: 14675–14684
Hu WH, Johnson H and Shu HB (2000) Activation of NF-kappaB by FADD, Casper, and Caspase-8. J. Biol. Chem. 275: 10838–10844
Ravi R, Bedi A, Fuchs EJ and Bedi A (1998) CD95 (Fas)-induced caspase-mediated proteolysis of NF-kappaB. Cancer Res. 58: 882–886
Levkau B, Scatena M, Giachelli CM et al. (1999) Apoptosis overrides survival signals through a caspase-mediated dominant-negative NF-kappa B loop. Nat. Cell Biol. 1: 227–233
Bachelder RE, Ribick MJ, Marchetti A et al. (1999) p53 inhibits alpha 6 beta 4 integrin survival signaling by promoting the caspase 3-dependent cleavage of AKT/PKB. J. Cell Biol. 147: 1063–1072
Yang DD, Kuan CY, Whitmarsh AJ et al. (1997) Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 389: 865–870
Behrens A, Sibilia M and Wagner EF (1999) Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 21: 326–329
Tournier C, Hess P, Yang DD et al. (2000) Requirement of JNK for stress-induced activation of the cytochrome c- mediated death pathway. Science 288: 870–874
Maundrell K, Antonsson B, Magnenat E et al. (1997) Bcl-2 undergoes phosphorylation by c-Jun N-terminal kinase/stress- activated protein kinases in the presence of the constitutively active GTP-binding protein Rac1. J. Biol. Chem. 272: 25238–25242
Yamamoto K, Ichijo H and Korsmeyer SJ (1999) BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell Biol. 19: 8469–8478
Srivastava RK, Mi QS, Hardwick JM and Longo DL (1999) Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc. Natl. Acad. Sci. USA 96: 3775–3780
Kuan CY, Yang DD, Samanta Roy DR et al. (1999) The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 22: 667–676
Nishina H, Fischer KD, Radvanyi L et al. (1997) Stress-signalling kinase Sek1 protects thymocytes from apoptosis mediated by CD95 and CD3. Nature 385: 350–353
Hochedlinger K, Wagner EF and Sabapathy K (2002) Differential effects of JNK1 and JNK2 on signal specific induction of apoptosis. Oncogene 21: 2441–2445
Liu H, Jones BE, Bradham C and Czaja MJ (2002) Increased cytochrome P-450 2E1 expression sensitizes hepatocytes to c-Jun-mediated cell death from TNF-alpha. Am. J. Physiol. Gastrointest. Liver Physiol. 282: G257–G266
Liu H, Lo CR and Czaja MJ (2002) NF-kappaB inhibition sensitizes hepatocytes to TNF-induced apoptosis through a sustained activation of JNK and c-Jun. Hepatology 35: 772–778
Guo YL, Baysal K, Kang B et al. (1998) Correlation between sustained c-Jun N-terminal protein kinase activation and apoptosis induced by tumor necrosis factor-alpha in rat mesangial cells. J. Biol. Chem. 273: 4027–4034
Guo YL, Kang B and Williamson JR (1998) Inhibition of the expression of mitogen-activated protein phosphatase-1 potentiates apoptosis induced by tumor necrosis factor-alpha in rat mesangial cells. J. Biol. Chem. 273: 10362–10366
Gabai VL, Mabuchi K, Mosser DD and Sherman MY (2002) Hsp72 and stress kinase c-jun N-terminal kinase regulate the bid- dependent pathway in tumor necrosis factor-induced apoptosis. Mol. Cell Biol. 22: 3415–3424
Tang G, Minemoto Y, Dibling B et al. (2001) Inhibition of JNK activation through NF-kappaB target genes. Nature 414: 313–317
Javelaud D and Besancon F (2001) NF-kappa B activation results in rapid inactivation of JNK in TNF alpha-treated Ewing sarcoma cells: a mechanism for the anti-apoptotic effect of NF-kappa B. Oncogene 20: 4365–4372
Muhlenbeck F, Haas E, Schwenzer R et al. (1998) TRAIL/Apo2L activates c-Jun NH2-terminal kinase (JNK) via caspase- dependent and caspase-independent pathways. J. Biol. Chem. 273: 33091–33098
Roulston A, Reinhard C, Amiri P and Williams LT (1998) Early activation of c-Jun N-terminal kinase and p38 kinase regulate cell survival in response to tumor necrosis factor alpha. J. Biol. Chem. 273: 10232–10239
Lenczowski JM, Dominguez L, Eder AM et al. (1997) Lack of a role for Jun kinase and AP-1 in Fas-induced apoptosis. Mol. Cell Biol. 17: 170–181
Herr I, Wilhelm D, Meyer E et al. (1999) JNK/SAPK activity contributes to TRAIL-induced apoptosis. Cell Death Differ. 6: 130–135
Geleziunas R, Xu W, Takeda K et al. (2001) HIV-1 Nef inhibits ASK1-dependent death signalling providing a potential mechanism for protecting the infected host cell. Nature 410: 834–838
Hatai T, Matsuzawa A, Inoshita S et al. (2000) Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J. Biol. Chem. 275: 26576–26581
Charette SJ, Lambert H, Landry J (2001) A kinase-independent function of Ask1 in caspase-independent cell death. J. Biol. Chem. 276: 36071–36074
De Smaele E, Zazzeroni F, Papa S et al. (2001) Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature 414: 308–313
Sulciner DJ, Irani K, Yu ZX et al. (1996) rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappaB activation. Mol. Cell Biol. 16: 7115–7121
Schulze-Osthoff K, Beyaert R, Vandevoorde V et al. (1993) Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene-inductive effects of TNF. EMBO J. 12: 3095–3104
Moreno-Manzano V, Ishikawa Y, Lucio-Cazana J and Kitamura M (2000) Selective involvement of superoxide anion, but not downstream compounds hydrogen peroxide and peroxynitrite, in tumor necrosis factor-alpha- induced apoptosis of rat mesangial cells. J. Biol. Chem. 275: 12684–12691
Binder C, Schulz M, Hiddemann W and Oellerich M (1999) Induction of inducible nitric oxide synthase is an essential part of tumor necrosis factor-alpha-induced apoptosis in MCF-7 and other epithelial tumor cells. Lab. Invest. 79: 1703–1712
Sidoti-de Fraisse C, Rincheval V, Risler Y et al. (1998) TNF-alpha activates at least two apoptotic signaling cascades. Oncogene 17: 1639–1651
Wong GH, Elwell JH, Oberley LW and Goeddel DV (1989) Manganous superoxide dismutase is essential for cellular resistance to cytotoxicity of tumor necrosis factor. Cell 58: 923–931
Wong GH and Goeddel DV (1988) Induction of manganous superoxide dismutase by tumor necrosis factor: possible protective mechanism. Science 242: 941–944
Leist M and Jaattela M (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2: 589–598
Holler N, Zaru R, Micheau O et al. (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1: 489–495
Luschen S, Ussat S, Scherer G et al. (2000) Sensitization to death receptor cytotoxicity by inhibition of fas- associated death domain protein (FADD)/caspase signaling. Requirement of cell cycle progression. J. Biol. Chem. 275: 24670–24678
Khwaja A and Tatton L (1999) Resistance to the cytotoxic effects of tumor necrosis factor alpha can be overcome by inhibition of a FADD/caspase-dependent signaling pathway. J. Biol. Chem. 274: 36817–36823
Vercammen D, Brouckaert G, Denecker G et al. (1998) Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med. 188: 919–930
Li M and Beg AA (2000) Induction of necrotic-like cell death by tumor necrosis factor alpha and caspase inhibitors: novel mechanism for killing virus-infected cells. J. Virol. 74: 7470–7477
Vercammen D, Beyaert R, Denecker G et al. (1998) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 187: 1477–1485
Kawahara A, Ohsawa Y, Matsumura H et al. (1998) Caspase-independent cell killing by Fas-associated protein with death domain. J. Cell Biol. 143: 1353–1360
Hennet T, Bertoni G, Richter C and Peterhans E (1993) Expression of BCL-2 protein enhances the survival of mouse fibrosarcoid cells in tumor necrosis factor-mediated cytotoxicity. Cancer Res. 53: 1456–1460
Hennet T, Richter C and Peterhans E (1993) Tumour necrosis factor-alpha induces superoxide anion generation in mitochondria of L929 cells. Biochem. J. 289: 587–592
Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B et al. (1992) Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J. Biol. Chem. 267: 5317–5323
Leist M, Single B, Castoldi AF et al. (1997) Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med. 185: 1481–1486
Eguchi Y, Shimizu S and Tsujimoto Y (1997) Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 57: 1835–1840
Ferrari D, Stepczynska A, Los M et al. (1998) Differential regulation and ATP requirement for caspase-8 and caspase-3 activation during. J. Exp. Med. 188: 979–984
Ha HC and Snyder SH (1999) Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 96: 13978–13982
D'Amours D, Sallmann FR, Dixit VM and Poirier GG (2001) Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: implications for apoptosis. J. Cell Sci. 114: 3771–3778
Los M, Mozoluk M, Ferrari D et al. (2002) Activation and Caspase-mediated inhibition of PARP: A molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol. Biol. Cell 13: 978–988
Schulze-Osthoff K, Krammer PH and Droge W (1994) Divergent signalling via APO-1/Fas and the TNF receptor, two homologous molecules involved in physiological cell death. EMBO J. 13: 4587–4596
Obeid LM, Linardic CM, Karolak LA and Hannun YA (1993) Programmed cell death induced by ceramide. Science 259: 1769–1771
Wiegmann K, Schutze S, Machleidt T et al. (1994) Functional dichotomy of neutral and acidic sphingomyelinases in tumor necrosis factor signaling. Cell 78: 1005–1015
Dressler KA, Mathias S and Kolesnick RN (1992) Tumor necrosis factor-alpha activates the sphingomyelin signal transduction pathway in a cell-free system. Science 255: 1715–1718
Stoffel W (1999) Functional analysis of acid and neutral sphingomyelinases in vitro and in vivo. Chem. Phys. Lipids 102: 107–121
Lozano J, Menendez S, Morales A et al. (2001) Cell autonomous apoptosis defects in acid sphingomyelinase knockout fibroblasts. J. Biol. Chem. 276: 442–448
Strelow A, Bernardo K, Adam-Klages S et al. (2000) Overexpression of acid ceramidase protects from tumor necrosis factor-induced cell death. J. Exp. Med. 192: 601–612
Wiegmann K, Schwandner R, Krut O et al. (1999) Requirement of FADD for tumor necrosis factor-induced activation of acid sphingomyelinase. J. Biol. Chem. 274: 5267–5270
Schwandner R, Wiegmann K, Bernardo K et al. (1998) TNF receptor death domain-associated proteins TRADD and FADD signal activation of acid sphingomyelinase. J. Biol. Chem. 273: 5916–5922
Heinrich M, Wickel M, Schneider-Brachert W et al. (1999) Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J. 18: 5252–5263
Bursch W (2001) The autophagosomal–lysosomal compartment in programmed cell death. Cell Death Differ. 8: 569–581
Stoka V, Turk B, Schendel SL et al. (2001) Lysosomal protease pathways to apoptosis. Cleavage of bid, not pro-caspases, is the most likely route. J. Biol. Chem. 276: 3149–3157
Foghsgaard L, Wissing D, Mauch D et al. (2001) Cathepsin B acts as a dominant execution protease in tumor cell apoptosis induced by tumor necrosis factor. J. Cell Biol. 153: 999–1010
Guicciardi ME, Deussing J, Miyoshi H et al. (2000) Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Invest. 106: 1127–1137
Deiss LP, Galinka H, Berissi H et al. (1996) Cathepsin D protease mediates programmed cell death induced by interferon-gamma, Fas/APO-1 and TNF-alpha. EMBO J. 15: 3861–3870
Guicciardi ME, Miyoshi H, Bronk SF and Gores GJ (2001) Cathepsin B knockout mice are resistant to tumor necrosis factor-alpha-mediated hepatocyte apoptosis and liver injury: implications for therapeutic applications. Am. J. Pathol. 159: 2045–2054
Heller RA, Song K, Fan N and Chang DJ (1992) The p70 tumor necrosis factor receptor mediates cytotoxicity. Cell 70: 47–56
Tartaglia LA, Rothe M, Hu YF and Goeddel DV (1993) Tumor necrosis factor's cytotoxic activity is signaled by the p55 TNF receptor. Cell 73: 213–216
Vandenabeele P, Declercq W, Vanhaesebroeck B et al. (1995) Both TNF receptors are required for TNF-mediated induction of apoptosis in PC60 cells. J. Immunol. 154: 2904–2913
Bigda J, Beletsky I, Brakebusch C et al. (1994) Dual role of the p75 tumor necrosis factor (TNF) receptor in TNF cytotoxicity. J. Exp. Med. 180: 445–460
Grell M, Scheurich P, Meager A and Pfizenmaier K (1993) TR60 and TR80 tumor necrosis factor (TNF)-receptors can independently mediate cytolysis. Lymphokine Cytokine Res. 12: 143–148
Medvedev AE, Sundan A and Espevik T (1994) Involvement of the tumor necrosis factor receptor p75 in mediating cytotoxicity and gene regulating activities. Eur. J. Immunol. 24: 2842–2849
Grell M, Zimmermann G, Gottfried E et al. (1999) Induction of cell death by tumour necrosis factor (TNF) receptor 2, CD40 and CD30: a role for TNF-R1 activation by endogenous membrane-anchored TNF. EMBO J 18: 3034–3043
Vercammen D, Vandenabeele P, Declercq W et al. (1995) Cytotoxicity in L929 murine fibrosarcoma cells after triggering of transfected human p75 tumour necrosis factor (TNF) receptor is mediated by endogenous murine TNF. Cytokine 7: 463–470
Schneider P, Schwenzer R, Haas E et al. (1999) TWEAK can induce cell death via endogenous TNF and TNF receptor 1. Eur. J. Immunol. 29: 1785–1792
Eliopoulos AG, Davies C, Knox PG et al. (2000) CD40 induces apoptosis in carcinoma cells through activation of cytotoxic ligands of the tumor necrosis factor superfamily. Mol. Cell Biol 20: 5503–5515
Yeh WC, Itie A, Elia AJ et al. (2000) Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 12: 633–642
Weitzman JB, Fiette L, Matsuo K and Yaniv M (2000) JunD protects cells from p53-dependent senescence and apoptosis. Mol. Cell 6: 1109–1119
Li ZW, Chu W, Hu Y et al. (1999) The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J. Exp. Med 189: 1839–1845
Senftleben U, Li ZW, Baud V and Karin M (2001) IKKbeta is essential for protecting T cells from TNFalpha-induced apoptosis. Immunity 14: 217–230
Yin L, Wu L, Wesche H et al. (2001) Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science 291: 2162–2165
Wang ZQ, Stingl L, Morrison C et al. (1997) PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 11: 2347–2358
Beg AA, Sha WC, Bronson RT et al. (1995) Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature 376: 167–170
Beg AA and Baltimore D (1996) An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 274: 782–784
Tsitsikov EN, Laouini D, Dunn IF et al. (2001) TRAF1 is a negative regulator of TNF signaling. Enhanced TNF signaling in TRAF1-deficient mice. Immunity 15: 647–657
Nakano H, Sakon S, Koseki H et al. (1999) Targeted disruption of TRAF5 gene causes defects in CD40- and CD27-mediated lymphocyte activation. Proc. Natl. Acad. Sci. USA 96: 9803–9808
Tada K, Okazaki T, Sakon S et al. (2001) Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF- kappa B activation and protection from cell death. J. Biol. Chem. 276: 36530–36534
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
Wajant, H., Pfizenmaier, K. & Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ 10, 45–65 (2003). https://doi.org/10.1038/sj.cdd.4401189
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401189
Keywords
This article is cited by
-
No-ozone cold plasma induces apoptosis in human neuroblastoma cell line via increased intracellular reactive oxygen species (ROS)
BMC Complementary Medicine and Therapies (2024)
-
Peptide Derivatives of Human and Rabbit Cathelicidin Reduce Inflammatory Cytokines in Peripheral Blood Mononuclear Cells of Rheumatoid Arthritis Patients
International Journal of Peptide Research and Therapeutics (2024)
-
Spirulina in fish immunity development: find the black box
Reviews in Fish Biology and Fisheries (2024)
-
The emerging role of glycolysis and immune evasion in gastric cancer
Cancer Cell International (2023)
-
Potent hepatoprotective activity of common rattan (Calamus rotang L.) leaf extract and its molecular mechanism
BMC Complementary Medicine and Therapies (2023)
