
Abstract
The IPC-81 myeloid leukaemia cells undergo apoptosis rapidly after cAMP stimulation (6 h) and cell death is prevented by early over-expression of the cAMP-inducible transcription repressor ICER, that blocks cAMP-dependent nuclear signalling. Therefore, the expression of specific genes controlled by CRE-containing promoters is likely to determine cell fate. We now show that cAMP-induced cell death also is abrogated by the over-expression of the anti-apoptotic gene, Bcl-2. Contrary to ICER, Bcl-2 does not affect cAMP-signalling and allows the analysis of cAMP responses in death rescued cells. The Bcl-2 transfected cells treated with 8-CPT-cAMP were growth-arrested and thereafter cells embarked in granulocytic differentiation, with no additional stimulation. Neutrophilic polynuclear granulocytes benefited from a long life span in G0-G1 and remained functional (phagocytosis). This work demonstrates that, using anti-apoptosis regulators, ‘death signals’ could be exploited to trigger distinct biological responses. Indeed, cAMP signal can trigger several simultaneously developing biological programs, in the same cell, i.e., growth regulation, apoptosis and differentiation. This cell system should prove useful to determine how a tumour cell can be re-programmed for either apoptosis or functional maturation by physiological signals. Cell Death and Differentiation (2000) 7, 1081–1089.
Similar content being viewed by others


Introduction
Cell homeostasis in higher organisms results from a subtle equilibrium between cell proliferation, differentiation and cell death. Programmed cell death (or apoptosis) plays an essential role in the ontogeny and function of normal cells, as well as in the development of tumours.1,2,3,4,5 Cell death can be read as the ultimate event of the differentiation program by which functionally mature cells are generated. In most tissues, fully differentiated cells function for a limited time and then undergo apoptosis. To avoid tissue ageing and ensure optimal renewal, differentiation and cell death should be tightly coupled programs. This notion is corroborated by analysis of normal haemopoietic cell differentiation where functionally mature cells, like mature polynuclear granulocytes, are short lived and die by apoptosis without additional signalling.6
It is well known that regulation of apoptosis occurs through control of pro- or anti-apoptotic genes, among which the genes of the Bcl-2 family are the best described (reviewed in7,8,9). Numerous studies have revealed a key role for Bcl-2 in the development of many tissues, including lymphoid cells, neuronal cells, epithelial cells of glandular tissues (for reviews see10,11,12,13). Although the mechanism(s) of action of the Bcl-2 protein and its partners remain(s) unclear, their transcriptional and post-transcriptional regulations support the hypothesis that these regulators might constitute molecular targets for several signalling pathways determining cell fate (reviewed in14). It is noteworthy that Bcl-2 down-regulation occurs during myeloid leukaemia cell differentiation.15,16,17,18 Bcl-2 and homologous anti-apoptotic proteins not only regulate cell survival, but control cell growth by an action at the cell cycle entry.19 However, Bcl-2 accomplishes these two distinct functions by genetically dissociated mechanisms.20 These findings not only suggest a diversification of functions for these proteins, but also that members of the Bcl-2 family may allow one signal to trigger more than one single biological response.
Cyclic-AMP elicits a ubiquitous signalling which plays a central role in diverse responses to hormone for cell proliferation, differentiation and apoptosis (reviewed in21,22). The current paradigm is that these distinct responses reflect either cell type related intrinsic differences, or signal cross-talks. While cAMP stimulates proliferation of some epitheliod cells,23 it induces differentiation in neuronal cells24,25,26,27,28 and apoptosis in thymocytes29 and B cell lymphomas.30 Alternatively, c-AMP synergises with glucocorticoids and PKC signalling in inducing apoptosis.28,29,31,32,33 In the case of human neutrophils several groups have reported that cAMP-elevating agents34,35 and cAMP analogues36 inhibited apoptosis. TGF-β and cAMP cooperate to promote apoptosis in B lymphocytes.37 Cyclic AMP also cooperates with nuclear receptor signalling to trigger cell differentiation. Cyclic-AMP and RARα-dependent signalling cross-talk results in the differentiation of F9 teratocarcinoma38,39,40,41 and granulocytic maturation of various leukaemia cells.42,43,44,45,46 Recently, a RARα-independent RXR signalling pathway has been evidenced by means of a RXR-PKA cross-talk necessary to induce maturation of retinoid-resistant leukaemia cells.47 In a specific cellular context, these signallings, taken either independently or combined, can trigger differentiation, proliferation and/or apoptosis. It has been convincingly established that triggering of these biological programs occurs through the transcriptional regulation of primary and secondary sets of genes. However, a pertinacious question that has to be answered is how one defined program is abrogated to allow the execution of another program.
Only a few in vitro biological systems offer the proper range of signals and biological responses to decipher the complex networks of molecular events that determine cell fate. The IPC-81 myeloid leukaemia cell line48 was used to study extensively the pathway for cAMP-induced apoptosis. Cyclic-AMP induces an early G1/S arrest49 and apoptosis with a complete cell destruction in a few hours.50,51,52 This apoptosis process requires PKA-dependent nuclear signalling (CREB phosphorylation) and can be abrogated by constitutive overexpression of the Inducible cAMP Early Repressor (ICER).53 Although ICER expressing cells were resistant to cAMP-induced cell death, they continued to proliferate with no sign of differentiation. Conceivably, the blockade of gene expression in these cells by constitutive ICER expression should not only block apoptosis-related genes, but also the expression of sets of genes involved in distinct CREB-dependent biological responses. In keeping with this hypothesis, it was of interest to determine whether cAMP could trigger additional biological responses, when only the early apoptotic response to cAMP stimulation was prevented. For this reason, we investigated whether the action of Bcl-2, which occurs downstream cAMP-dependent transcriptional regulation, could block apoptosis and thus allow the analysis of the biological response of death-rescued cells.
In this work, we show that using a physiological agent, a highly tumorigenic leukaemia cell line54 can be efficiently manipulated, either committed to death or reprogrammed for a normal process of morphological and functional maturation.
Results
Ectopic expression of Bcl-2 uncouples the death machinery from cAMP-dependent nuclear signalling
The MSCVpkg expression vector containing the Bcl-2 coding sequence under the control of the MSCV promoter,55 was used to produce ectopic retroviral particle in transiently transfected BOSC23 cells.56 The resulting virus preparations were used to infect both the IPC-81 and IPC-RID336 cells, respectively expressing the wild-type and a mutated regulatory subunit (RI) of the PKA.57 As experimental control, a similar ‘empty vector’ has also been used to transfect cells. Several cloned cell lines have been selected for puromycin resistance. To check for the retroviral integration of the Bcl-2 gene into IPC-81 genome and the ectopic expression of its mRNA, a human Bcl-2 cDNA was used in Southern and Northern blots analyses, respectively. A mouse monoclonal antibody directed against human Bcl-2 which cross-reacts with rat Bcl-2 was used for Western blot detection of both rat (endogenous) and human (ectopic) Bcl-2 proteins, comparatively (Figure 1).

IPC-81 cell transfection and over-expression of Bcl-2 mRNA and protein in IPC-81 and IPC-RID336 cells. (A) Southern blot analysis of Bcl-2 transfected IPC-81 cells. EcoRI, HindIII and BamHI DNA digests from IPC-81, IPC-e.v., IPC-Bcl-2, IPC-RID336 and IPC-RID336-Bcl-2 cells were hybridised with a human Bcl-2 probe. (B) Total mRNA from IPC-81, IPC-RID336 and transfected cells (IPC-e.v.; IPC-Bcl-2; IPC-RID336-Bcl-2) were analyzed comparatively on Northern blot for Bcl-2 mRNA expression. Note that in IPC-81 cells the endogenous Bcl-2 transcripts are hardly detectable on Northern-blot (endogenous bcl-2 mRNAs sized 7.5 kb, 4.1 kb, 1.9 kb, and therefore are not seen in this figure). (C) Total protein extracts from the same cells were analyzed by 15% SDS–PAGE. Bcl-2 proteins were detected by a anti-(rat/human) Bcl-2 mouse monoclonal antibody (see in Materials and Methods)
Bcl-2 transfected clones showed a positive hybridisation signal for ectopic Bcl-2, after EcoRI, HindIII and BamHI restriction enzyme digestion of DNA (Figure 1A). ‘Empty vector’ transfected cells (IPC-e.v.) also resistant to puromycin were modified neither in endogenous Bcl-2 expression, nor in the apoptotic response to cAMP or other agents inducing cell death as okadaic acid, calyculin A, FasL, doxorubicin (not shown). Bcl-2 transfectants (IPC-Bcl-2 and IPC-RI-Bcl-2) show a strong constitutive expression of a 5 kb mRNA (Figure 1B). Western-blot analyses of total cellular extracts indicate (Figure 1C) that Bcl-2 transfected cells also over-expressed a 26 kDa band corresponding to Bcl-2 protein. Thus, these transfected clones showed the presence of the transfected human Bcl-2 gene and the production of mRNAs and protein of expected sizes. These transfected cell clones over-expressing Bcl-2 showed no growth alteration (not shown).
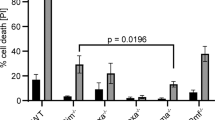
In order to compare IPC-e.v. and IPC-Bcl-2 transfectants for their response to cAMP, we used 8-CPT-cAMP, an analog which binds and activates PKA50 and induces apoptosis in IPC-81 cells, but not in the IPC-RID336 mutated cell line.57 Previous experiments on the IPC-81 cell line, showed that a 100 μM concentration of 8-CPT-cAMP induces apoptosis in more than 60% of the cell population after 4 h of incubation and that no cell survived after a 10 h treatment.50 In order to analyze the features of both IPC-Bcl-2 and IPC-e.v. cells, dose-response assays were performed. Cell viability assays revealed a marked shift in dose-response curves after 17 h of incubation (Figure 2A) showing that IPC-Bcl-2 transfected cells were protected against cAMP-induced cell death. The IC50 (35 μM for control cells) is shifted to more than 1000 μM for Bcl-2 transfectants. These results are in accordance with cell morphology (Figure 2B) and with agarose gel DNA fragmentation analysis (Figure 2C). DNA ladder, a typical feature for apoptotic cell death, is observed at 80 μM of 8-CPT-cAMP in control cells after only 5 h of cAMP treatment, whereas Bcl-2-transfected cells showed no DNA fragmentation. These experiments demonstrate for the first time that Bcl-2 over-expression protects these cells from PKA dependent apoptosis. To document further the suppression of cAMP-induced apoptosis by Bcl-2, caspase-3 like activation was evaluated after 5 h of treatment in IPC-e.v. and IPC-Bcl-2 cells, comparatively (Figure 2D). Clearly, no enzyme activity in cell extracts could cleave the caspase-3 like colorimetric substrate, Ac-DEVD-pNa. This feature was confirmed at later times during treatment (not shown). Altogether, these data indicated that ectopic over-expression of Bcl-2 in IPC cells, efficiently blocked cAMP-induced apoptosis.

Resistance of IPC-Bcl-2 cells to cAMP-induced apoptosis. (A) Cell viability was measured by WST1 assay after 17 h incubation of cultures with 8-CPT-cAMP. Dose-response to 8-CPT-cAMP (B). Cell morphology (May-Grünwald-Giemsa staining) after a 5 h 8-CPT-cAMP treatment (100 μM) of IPC-e.v. (B1) and IPC-Bcl-2 (B2) cells. (C) DNA fragmentation was evaluated by electrophoretic migration on agarose gels after 5 h incubation with 8-CPT-cAMP; Dose-effect of 8-CPT-cAMP on IPC-e.v. cells (left panel) and IPC-Bcl-2 cells (right panel). (D) Differential activation of caspase-3 like protease in IPC-e.v. and IPC-Bcl-2 by cAMP, estimated from the colorimetric measurement of Ac-DEVD-pNa cleavage in vitro. All experiments were carried out at least three times. Data shown are from a typical set of analyses
Rapid growth arrest induced by cAMP in Bcl-2 transfected cells
We wanted to determine the fate of cells rescued from death by Bcl-2 over-expression, particularly whether cAMP altered cell growth. Both IPC-e.v., and IPC-Bcl2 were examined for cell cycle and ‘TUNEL’ analyses during cAMP treatment (Figure 3).

Cell cycle analyses of IPC-e.v. and IPC-Bcl-2 cells by flow cytometry during 8-CPT-cAMP (100 μM) treatment. (A) Flow cytometry profiles. Apoptosis was evaluated by TUNEL reaction after 6 h of 8-CPT-cAMP treatment compared to control cultures. (B) kinetic of growth arrest versus apoptosis in IPC-Bcl-2 and IPC-e.v. cells treated by 8-CPT-cAMP (100 μM). All experiments were carried out at least three times. Data shown are from a typical set of analyses
In IPC-e.v. cells used in a control culture, 4 h after addition of 8-CPT-cAMP, a new peak was observed ahead of the G1 peak, on flow cytometry profiles. This additional pre-G1 peak corresponded to the accumulation of apoptotic cells.58 This peak increased in size after 6 h of cAMP treatment and ‘TUNEL’ reaction clearly showed a high level of apoptotic cells at the same time (Figure 3A). As expected, these flow cytometry profiles were similar to those obtained with the parental IPC-81 cell line receiving the same treatment (not shown). By contrast, no pre-G1 peak and labelling of DNA fragments could be observed for the Bcl-2-transfected cells, when analyzed after 6 h of cAMP treatments. This confirmed that cAMP-induced apoptosis was blocked in Bcl-2 transfected cells. Furthermore, it was clearly noticed that a decrease of the S-phase peak occurred as early as 6 h of cAMP-treatment (Figure 3A,B) and, remarkably, all cells were growth arrested after 16 h (within a lag-time corresponding to the average doubling-time of IPC-81 cells). By contrast, IPC-RID336-Bcl2 cells which bear a mutation in the R1 subunit of the PKA57 showed neither apoptosis nor growth inhibition by cAMP treatment (not shown). These results demonstrate that when Bcl-2 is over-expressed, the early apoptotic response is suppressed and, instead, growth inhibition by cAMP occurs 12–16 h later.
Fate of death-rescued IPC-Bcl-2 cells: recoupling of cAMP-dependent nuclear signalling to the granulocytic maturation program
We wanted to further analyze the fate of Bcl-2 rescued cells. Both viability curves and flow cytometry analyses concurred to support the conclusion that after 16 h of 8-CPT-cAMP treatment, all cells were growth arrested, most of them in G0-G1, a few being blocked in G2-M (Figure 3B). Morphological analysis (Figure 4A–C) carried out in parallel on growth arrested cells showed that after 72 h of cAMP treatment, IPC-Bcl-2 cells were achieving granulocytic maturation (Figure 4B). Granulocytic differentiation of IPC-Bcl-2 cells was then analyzed by flow cytometry, using specific differentiation markers such as the expression of membranous adhesion molecules, the CD11b and CD11c integrins. Figure 4D shows that from 24 to 96 h, Bcl-2 transfected cells treated with cAMP expressed increasing amounts of CD11b/c integrins. Therefore, not only Bcl2 expression rescued cells from death, and allowed growth arrest but, importantly, it allowed myeloid cell maturation to occur. Notice also that cAMP, which did not trigger apoptosis in the PKA-mutated IPC-RID336 cells,57 neither elicited maturation in the Bcl-2 transfected IPC-RID336-Bcl2 cells (not shown), further supporting the conclusion that PKA signalling triggers the two biological programs.

Morphological and functional granulocytic maturation of IPC-Bcl-2 cells in response to 8-CPT-cAMP (100 μM) treatment. May-Grünwald stained cultures, (A) control culture; (B) 8-CPT-cAMP treated cells (72 h) mature in polymorphonuclear granulocytes; (C) 8-CPT-cAMP treated cells (48 h) incubated with fixed E. coli and Saccharomyces cerevisae for 30 min. Note the typical morphology of mature polynuclear cells. Yeast and bacteria being engulfed by phagocytosis (arrows) and phagosome containing partially digested yeast (arrow head). (D) Flow cytometry analyses of CD11b/c integrin expression and yeast phagocytosis, during 8-CPT-cAMP treatment (100 μM). For phagocytosis activity, at least 100 cells were analyzed on each of four to six histological slides for each incubation. Data (positive cells) are expressed as % of the total cell population. All experiments were carried out at least three times. Data shown are from a typical set of analyses
Bcl-2 over-expression changes normally short-lived cells into long-lived functional polynuclear granulocytic phagocytes
It is well documented that normal granulocytic polynuclear phagocytes show no Bcl-2 expression. They are short-lived and the functional activity of these cells in vivo is generally limited to 10 to 15 h.59 These cells die by apoptosis, and macrophage recognition and phagocytosis occur before any release of their inflammatory content.6,60 cAMP signalling is also an important regulator for terminal maturation, chemotaxis and the death of these cells.34,35,36,61,62,63,64 We took advantage of cAMP-induced polynuclear cells to determine whether in leukaemia-derived granulocytes the unphysiological expression of Bcl-2 affected polynuclear phagocyte function and life span. Addition in cell culture medium of E. Coli and/or Saccharomyces cerevisiae previously killed by fixative treatments allowed the demonstration of an active phagocytosis by the cAMP-induced polynuclear cells, as early as 48 h after cAMP-induced maturation (Figure 4C). Prolonged cultures of the mature phagocytic cells indicated that cell survival of Bcl-2 transfected cells was maintained for up to 2 weeks in vitro. However, the function of phagocytes declined with time, then finally cells died.
Discussion
It is generally considered that cellular signalling pathways are univalent (i.e. death signalling, survival signalling, differentiation signalling). When one pathway signals for more than one biological response, the development of multiple responses is commonly understood as the consequence of variations in the intracellular components necessary for signal transduction. This notion could well explain why cAMP generates either growth stimulation, growth arrest, cell differentiation, cell death or inhibition of cell death in various hormone-dependent tissues. Considering that cAMP is a ubiquitous second messenger involved in regulation of gene expression, the involvement of CRE-dependent transcriptional regulation in determining cell fate is suspected.
In a previous work, we showed that in IPC-81 cells, cAMP-induced apoptosis depends on early gene expression, and is blocked by the ectopic expression of the cAMP inducible transcriptional repressor ICER.53 In this work, we show that, acting downstream the transcriptional regulation by cAMP, Bcl-2 efficiently protected IPC-81 cells from apoptosis. Importantly, although enhanced Bcl-2 expression prevented cAMP from inducing apoptosis, a rapid growth arrest occurred in the death-rescued cells after cAMP triggering, then granulocyte differentiation was observed. Noticeably, in the absence of cAMP treatment, the ectopic expression of Bcl-2 neither modified the cell cycle, nor triggered cell maturation.
Polynuclear neutrophil granulocytes developed in culture after cAMP signalling became fully mature and functional after only 48 h of cAMP stimulation. It is of interest that development of normal granulocytes from myelocytes, during normal haemopoiesis, requires a roughly similar time-course, but that fully mature cells downregulate Bcl-2, are short-lived and die by apoptosis a few hours after cell maturation. In IPC-Bcl-2 leukaemia cells, cAMP acts as a potent intracellular messenger for terminal granulocyte maturation. In these cells, not only does Bcl-2 not affect terminal granulocytic maturation but also it prolongs dramatically the life span of granulocytic phagocytes. Remarkedly, when constitutively expressed, Bcl-2 sequentially protects IPC-81 blast cells from early apoptosis, allows the development of cAMP-induced cell differentiation and increases the survival of terminally mature cells (Figure 5). Considering work reported by other groups34,35,36 showing that cAMP inhibited apoptosis of terminally mature human neutrophils, we cannot exclude that cAMP also contributed to a prolonged survival of cells rescued by Bcl-2 from apoptosis at the immature stage. If this feature was sustained by experimental data, it would mean that, when acting on the same cell type, cAMP can signal for terminal maturation, apoptosis and cell survival, depending upon the stage of cell differentiation. Broadly speaking, the cross-talk between pathways regulating Bcl-2 expression and/or activity and the various signalling for cell differentiation is likely to determine the command of tissue development and remodelling.

Schematical representation of the cAMP-dependent signalling pathway for cell death and differentiation. A cascade of proximal events (generation of cAMP, activation of PKA, CREB phosphorylation) is shared by death and differentiation signallings and blocked by ICER overexpression.53 By means of transcriptional gene regulation, cAMP signalling activates distinct sets of genes, each set of genes being necessary to the development of distinct programs, either early cell death of immature blast cells (P1) or myeloid differentiation, terminal maturation, and then death of mature cells (P2). P1 causing early cell death occurs before the development of the distal events of P2 necessary to differentiation. Bcl-2 over-expression represses cAMP-induced cell death without effect on development of P2 program towards cell maturation. Ectopic expression of Bcl-2 in mature cells prolongs their survival as functional phagocytes
Sustained expression of anti-apoptotic regulators like the Bcl-2 family members might be important to the normal development of myelopoiesis. They may provide a second level of regulation for cell survival, after haemopoietic growth factors which are essentially survival factors for haemopoietic progenitors. As a matter of fact, Bcl-2 promotes cell survival in factor-deprived cultures without stimulation of cell proliferation. Bcl-2 rescued cells are most often altered in cell proliferation, at cell cycle entry.19 However, it is not yet clear whether alteration resulted from a lack of intrinsic capacity to proliferate or from a direct action of Bcl-2 on the cell cycle.65 Other authors have suggested that Bcl-2 actions on cell survival and growth controls are likely achieved through distinct mechanisms.20,66 Bcl-2 over-expression suppresses cell death without effect on cell capacity to differentiate.67 In the case of human neutrophils, ectopic overexpression of Bcl-2 noticeably prolongs cell survival, but interestingly does not block all phenotypic changes preceding or accompanying cell death; indeed Bcl-2 does not prevent ageing neutrophils to be engulfed by macrophages.68 It has also been reported that death-rescued factor-dependent cells undergo spontaneous differentiation, suggesting intrinsic maturation potential.69 It is not clear whether this observation can be extended to other cell types (e.g. autonomous tumour cells, like IPC-81 cells), or whether death-rescued cells require appropriate regulatory signal(s) to differentiate. Indeed, this work has shown that in IPC-81 cells, cAMP signalling causing cell death and growth arrest is also required for cell maturation. Because numerous physiological signals known to be important players in haemopoietic differentiation (cAMP, retinoids, glucocorticoids, VD3) also trigger cell death, a sustained Bcl-2 expression (or expression of other members of the Bcl-2 family) could be required for haemopoietic cell development, particularly the full accomplishment of the granulocytic differentiation program. The death program could represent an ancillary program, made available at any time during differentiation, by only modulating the expression of anti-apoptotic regulators.
Finally, this cell model system should prove useful to further investigate to what extent programmed cell death and differentiation share a common signalling pathway and effectors, when activated physiologically. Our knowledge of the molecular network involved in coupling cell death and differentiation programs could benefit to novel therapeutical approaches of tumour treatments.
Materials and Methods
Cell culture and viability assay
The rat promyelocytic leukaemia cell line IPC-81 was previously derived from BN rat myeloid leukaemia (BNML)48 and cultured as previously described.50 Note that the IPC-81 cell line is a fully growth-autonomous tumour cell line, and thus it does not require survival factors (CSFs or interleukins) in culture media. Cell morphology was analyzed using the May-Grünwald-Giemsa staining. Cell viability was assessed by morphological integrity of cells under phase-contrast microscopy and by the WST-1 assay (Boehringer Mannheim).
Plasmids, cell transfection, selection of Bcl-2-transfected IPC-81 cells
The MSCVpkgBcl-2 vector was obtained by inserting the complete human Bcl-2 cDNA sequence10 into the EcoRI restriction site of a MSCVpkg vector containing a puromycin resistance gene.54 The constructs, MSCVpkgBcl-2 and MSCVpkg as control, were used to derive ecotropic retroviral particles in BOSC23 cells.56 Selection of stable transfected clones was performed by maintaining the culture at a 0.8 μg/ml puromycin concentration during 6 weeks. After amplification of surviving cells, proteins and RNA were analyzed on Western and Northern blots, respectively.
Southern blot and Northern blot analyses
DNA was extracted using standard protocol. After completed digestion with EcoRI, HindIII and BamHI restriction endonucleases, genomic DNA (10 μg) was electrophoresed, denatured and transferred onto nylon membranes (Hybond-N; Amersham) as described by Maniatis et al.70 and modified by Hillion et al.71 Total RNA was isolated according to the procedure described by Chomczynsky and Sacchi.72 Twenty μg of total RNA were electrophoresed in a 1.1% agarose/10% formaldehyde gel and then blotted on a nylon membrane.
DNA fragmentation
Cells were pelleted and immediately disrupted in lysis buffer (10 mM Tris-HCl pH 8, 100 mM EDTA, 10 mM EGTA, 0.5% SDS), according to Williams et al.73 DNase-free RNase (SIGMA) was added at 20 μg/ml to the lysates, which were incubated for 2 h at 37°C. The cell lysates were then incubated at 56°C with proteinase K at 100 mg/ml for 1 h. The DNA was extracted with phenol, pelleted with 2 volumes ethanol and 0.1 volume 10 M ammonium acetate, dissolved in TE-buffer (10 mM Tris-HCl pH 8, 1 mM EDTA) and separated on a 1.5% agarose gel (FMC Bioproducts, USA). DNA fragments were visualised on gels after ethidium bromide staining.
Measurement of caspase-3 activity
Caspase-3 like activity was measured using a DEVD cleavage assay modified from Wright et al74 in which DEVD-pNA was used as a colorimetric substrate peptide (Biomol, Tebu, France). Briefly, cells (2×106) were pelleted, washed with PBS, pH 7.2, and lysed in 50 mM Tris-HCl, pH 7.5, 0.03% NP-40, 1.0 mM DTT. Lysates were centrifuged at 14 000 r.p.m., for 15 min at 4°C. Total protein determination was done using the Bradford assay. Assays were set up in flat bottom 96-well plates containing 0.2 mM of Ac-DEVD-pNA in a caspase reaction buffer (100 mM HEPES, pH 7.5, 10% sucrose, 0.1% CHAPS, 10 mM DTT) and 0.01 ml of protein extract (20–50 μg) in a total volume of 0.1 ml. Assays were incubated at 37°C. Release of pNa was detected by periodic readings of absorbance at 405 nm taken against a blank containing buffer and peptide alone (i.e., no extract) from 0–5 h to mark the linearity of the enzymatic reaction in time. Enzyme activities were measured as initial velocities and expressed as relative intensity/min/mg total protein within the linear range of the response.
Western blot analysis of protein expression
Cultured cells were washed in PBS and pelleted by centrifugation at 400×g for 5 min. Pellets of 5×105 cells were immediately lysed by adding 100 μl of a boiling Laemmli solution containing β-mercaptoethanol and disrupted with a pestle. Samples were then boiled for 5 min and insoluble material removed by centrifugation at 13 000×g for 5 min. Protein extracts (30 μg) were loaded on 15% SDS–polyacrylamide gel, electrophoresed, and blotted onto nitrocellulose membranes (Schleicher & Schuell, Germany). After transfer to the nitrocellulose membrane, proteins were visualised with Ponceau S (Sigma) to confirm equal loading of protein. Membranes were blocked with 5% unfatted milk in PBS, then incubated with a mouse monoclonal antiserum (1/500) raised against human Bcl-2 protein (Dako) in PBS/3% milk for 18 h at 4°C. Membranes were then incubated with horseradish peroxidase-linked protein A for 30 min at 25°C. Each of these steps were followed by three washes for 10 min in PBS/3% milk. Labelling was performed as described in the ECL protocol (Amersham).
Flow cytometry analysis (FACS) of cell surface antigens and cell cycle
The expression of the membrane adhesion molecules CD11b and c integrins was analyzed by direct immuno-fluorescence. After 8-CPT-cAMP (100 μM) incubations, cells were washed in PBS and labelled with an anti-rat CD11b-c FITC mouse monoclonal antibody (Caltag Labs). Cells were then washed twice in PBS and fixed in 1% paraformaldehyde/PBS solution. Cells were analyzed using an FACSCALIBUR (Becton-Dickinson) flow-cytometer. For cell cycle analysis, cells were washed in serum-free medium then fixed in 70% ethanol.
Abbreviations
- cAMP:
-
cyclic adenosine monophosphate
- 8-CPT-cAMP:
-
8-chloro-phenyl-thio-cAMP
- CRE:
-
c-AMP responsive element
- FACS:
-
fluorescence-activated cell sorting
- PKC:
-
Protein kinase C
- PKA:
-
c-AMP-dependent protein kinase
- RAR:
-
retinoic acid receptor
- RXR:
-
retinoid-X-receptor
- TUNEL:
-
terminal deoxynucleotide transferase-mediated dUTP-biotin nick-end labelling
References
Arends MJ and Wyllie AH . 1991 Apoptosis: mechanisms and roles in pathology. Int. Rev. Exp. Pathol. 32: 223–254
Fisher DE . 1994 Apoptosis in cancer therapy: crossing the threshold. Cell 78: 539–542
Hoffman B and Liebermann DA . 1994 Molecular controls of apoptosis: differentiation/growth arrest primary response genes, proto-oncogenes, and tumor suppressor genes as positive and negative modulators. Oncogene 9: 1807–1812
White E . 1996 Life, death, and the pursuit of apoptosis. Genes Dev. 10: 1–15
Rowan S and Fisher DE . 1997 Mechanisms of apoptotic cell death. Leukemia 11: 457–465
Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM and Haslett C . 1989 Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Invest. 83: 865–875
Zamzami N, Brenner C, Marzo I, Susin SA and Kroemer G . 1998 Subcellular and submitochondrial mode of action of Bcl-2-like oncoproteins. Oncogene 16: 2265–2282
Reed J . 1998 Bcl-2 family proteins. Oncogene 17: 3225–3236
Chao DT and Korsmeyer SJ . 1998 Bcl-2 family: Regulators of Cell Death. Annu. Rev. Immunol. 16: 395–419
Hockenbery DM . 1994 Bcl-2 in cancer, development and apoptosis. J. Cell. Sci. (Suppl.) 18: 51–55
Cory S, Harris AW and Strasser A . 1994 Insights from transgenic mice regarding the role of bcl-2 in normal and neoplastic lymphoid cells. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 345: 289–295
Merry DE and Korsmeyer SJ . 1997 Bcl-2 gene family in the nervous system. Annu. Rev. Neurosci. 20: 245–267
Hawkins CJ and Vaux DL . 1997 The role of the Bcl-2 family of apoptosis regulatory proteins in the immune system. Semin. Immunol. 9: 25–33
Dragovich T, Rudin CM and Thompson CB . 1998 Signal transduction pathways that regulate cell survival and cell death. Oncogene 17: 3207–3213
Naumovski L and Cleary ML . 1994 Bcl2 inhibits apoptosis associated with terminal differentiation of HL-60 myeloid leukemia cells. Blood 83: 2261–2267
Bruel A, Benoit G, De Nay D, Brown S and Lanotte M . 1995 Distinct apoptotic responses in maturation sensitive and resistant t(15;17) acute promyelocytic leukemia NB4 cells. 9-cis retinoic acid induces apoptosis independent of maturation and Bcl-2 expression. Leukemia 9: 1173–1184
Nagy L, Thomazy VA, Shipley GL, Fesus L, Lamph W, Heyman RA, Chandraratna RA and Davies PJ . 1995 Activation of retinoid X receptors induces apoptosis in HL-60 cell lines. Mol. Cell. Biol. 15: 3540–3551
Grillier I, Umiel T, Elstner E, Collins SJ and Koeffler HP . 1997 Alterations of differentiation, clonal proliferation, cell cycle progression and bcl-2 expression in RAR alpha-altered sublines of HL-60. Leukemia 11: 393–400
O'Reilly LA, Huang DC and Strasser A . 1996 The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J. 15: 6979–6990
Huang DC, O'Reilly LA, Strasser A and Cory S . 1997 The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J. 16: 4628–4638
Spaulding SW . 1993 The ways in which hormones change cyclic adenosine 3′,5′-monophosphate-dependent protein kinase subunits, and how such changes affect cell behavior. Endocr. Rev. 14: 632–650
Gjertsen B and Doskeland SO . 1995 Protein phosphorylation in apoptosis. Biochim. Biophys. Acta 1269: 187–199
Dumont J, Jauniaux JC and Roger PP . 1989 The cyclic AMP-mediated stimulation of cell proliferation. Trends Biochem. Sci. 14: 67–71
Braumann T, Jastorff B and Richter-Landsberg C . 1986 Fate of cyclic nucleotides in PC12 cell cultures: uptake, metabolism, and effects of metabolites on nerve growth factor-induced neurite outgrowth. J. Neurochem. 47: 912–919
Nicolai S, Willems J, Zwijsen A, Van Mechelen E and Slegers H . 1996 Cyclic AMP-induced differentiation increases the synthesis of extracellular superoxide dismutase in rat C6 glioma. Free Radic. Biol. Med. 21: 481–486
Sharma SK and Raj AB . 1987 Transient increase in intracellular concentration of adenosine 3′ : 5′-cyclic monophosphate results in morphological and biochemical differentiation of C6 glioma cells in culture. J. Neurosci. Res. 17: 135–141
Ginty DD, Fanger GR, Wagner JA and Maue RA . 1992 The activity of cAMP-dependent protein kinase is required at a posttranslational level for induction of voltage-dependent sodium channels by peptide growth factors in PC12 cells. J. Cell Biol. 116: 1465–1473
Kvanta A and Fredholm BB . 1993 Synergistic effects between protein kinase C and cAMP on activator protein-1 activity and differentiation of PC-12 pheocyromocytoma cells. J. Mol. Neurosci. 4: 205–214
McConkey DJ, Orrenius S, Okret S and Jondal M . 1993 Cyclic AMP potentiates glucocorticoid-induced endogenous endonuclease activation in thymocytes. FASEB J. 7: 580–588
Brown D and Phipps RP . 1996 Bcl-2 expression inhibits prostaglandin E2-mediated apoptosis in B cell lymphomas. J. Immunol. 157: 1359–1370
Suzuki K, Kizaki H, Tadakuma T and Ishimura Y. . 1990 12-O-tetradecanoylphorbol 13-acetate potentiates the action of cAMP in inducing DNA cleavage in thymocytes. Biochem. Biophys. Res. Commun. 171: 827–831
Gruol D and Altschmied J . 1993 Synergistic induction of apoptosis with glucocorticoids and 3′, 5′-cyclic adenosine monophosphate reveals agonist activity RU 486. Mol. Endocrinol. 7: 104–113
Dowd D and Miesfeld RL . 1992 Evidence that glucocorticoid- and cyclic AMP-induced apoptotic pathways in lymphocytes share distal events. Mol. Cell Biol. 12: 3600–3608
Parvathenami LK, Buescher ES, Chacon-Cruz E and Beebe SJ . 1998 Type 1 cAMP-dependent protein kinase delays apoptosis in Human Neutrophils at a site upstream of caspase-3. J. Biol. Chem. 273: 6736–6743
Ottonello L, Gonella R, Dapino P, Sacchetti C and Dallegri F . 1998 Prostaglandin E2 inhibits apoptosis in human neutrophilic polymorphonuclear leukocytes: Role of intracellular cyclic AMP levels. Exp. Hematol. 26: 895–902
Rossi AG, Cousin JM, Dransfield I, Lawson MF, Chilvers ER and Haslett C . 1995 Agents that elevate cAMP inhibit human neutrophil apoptosis. Biochem. Biophys. Res. Commun. 217: 892–899
Lomo J, Blomhoff HK, Beiske K, Stokke T and Smeland EB . 1995 TGF-beta 1 and cyclic AMP promote apoptosis in resting human B lymphocytes. J. Immunol. 154: 1634–1643
Goldstein B, Rogelj S, Siegel S, Farmer SR and Niles RM . 1990 Cyclic adenosine monophosphate-mediated induction of F9 teratocarcinoma differentiation in the absence of retinoic acid. J. Cell Physiol. 143: 205–212
Martin CA, Ziegler LM and Napoli JL . 1990 Retinoic acid, dibutyryl-cAMP, and differentiation affect the expression of retinoic acid receptors in F9 cells. Proc. Natl. Acad. Sci. U.S.A. 87: 4804–4808
Damjanov I, Zhu ZM, Andrews PW and Fenderson BA . 1994 Embryonal carcinoma cells differentiate into parietal endoderm via an intermediate stage corresponding to primitive endoderm. In Vivo 8: 967–973
Taneja R, Rochette-Egly C, Plassat JL, Penna L, Gaub MP and Chambon P . 1997 Phosphorylation of activation functions AF-1 and AF-2 of RAR alpha and RAR gamma is indispensable for differentiation of F9 cells upon retinoic acid and cAMP treatment. EMBO J. 16: 6452–6465
Olsson IL, Breitman TR and Gallo RC . 1982 Priming of human myeloid leukemic cell lines HL-60 and U-937 with retinoic acid for differentiation effects of cyclic adenosine 3′ : 5′-monophosphate-inducing agents and a T-lymphocyte-derived differentiation factor. Cancer Res. 42: 3928–3933
Ruchaud S, Duprez E, Gendron MC, Houge G, Genieser HG, Jastorff B, Doskeland SO and Lanotte M . 1994 Two distinctly regulated events, priming and triggering, during retinoid-induced maturation and resistance of NB4 promyleocytic leukemia cell line. Proc. Natl. acad. Sci. U.S.A. 91: 8428–8432
Gianni M, Terao M, Norio P, Barbui T, Rambaldi A and Garattini E . 1995 All-trans retinoic acid and cyclic adenosine monophosphate cooperate in the expression of leukocyte alkaline phosphatase in acute promyleocytic leukemia cells. Blood 85: 3619–3635
Chen A, Licht JD, Wu Y, Hellinger N, Scher W and Waxman S . 1994 Retinoic acid is required for and potentiates differentiation of acute promyelocytic leukemia cells by nonretinoid agents. Blood 84: 2122–2129
Quenech'Du N, Ruchaud S, Khelef N, Guiso N and Lanotte M . 1998 A sustained increase in the endogenous level of cAMP reduces the retinoid concentration required for APL cell maturation to near physiological levels. Leukemia 12: 1829–1833
Benoit G, Altucci L, Flexor M, Ruchaud S, Lillehaug J, Raffelsberger W, Gronemeyer H and Lanotte M . 1999 RAR-independent RXR signaling induces t(15;17) leukemia cell maturation. EMBO J. 18: 7011–7018
Lanotte M, Hermouet S, Gombaud-Saintonge G and Dobo I . 1986 On growth regulation of the rat promyelocytic leukemia (BNML): growth inhibition and eradication of clonogenic cells by cholera toxin. Leuk. Res. 10: 1319–1326
Gjertsen BT, Cressey LI, Ruchaud S, Houge G, Lanotte M and Doskeland SO . 1994 Multiple apoptotic death types triggered through activation of separate pathways by cAMP and inhibitors of protein phosphatases in one (IPC leukemia) cell line. J. Cell Sci. 107: 3363–3377
Lanotte M, Riviere JB, Hermouet S, Houge G, Vintermyr OK, Gjertsen BT and Doskeland SO . 1991 Programmed cell death (apoptosis) is induced rapidly and with positive cooperativity by activation of cyclic adenosine monophosphate-kinase I in a myeloid leukemia cell line. J. Cell Physiol. 146: 73–80
Vintermyr O, Gjertsen BT, Lanotte M and Doskeland SO . 1993 Microinjected catalytic subunit of cAMP-dependent protein kinase induces apoptosis in myeloid leukemia (IPC-81) cells. Exp. Cell. Res. 206: 157–161
Ruchaud S, Zorn M, Winkler E, Genieser HG, Doskeland SO, Jastorff B and Lanotte M . 1995 Evidence for several pathways of biological responses to hydrolysable cAMP-analogs using a model system of apoptosis. Cell. Pharmacol. 2: 127–140
Ruchaud S, Seite P, Foulkes NS, Sassone-Corsi P and Lanotte M . 1997 The transcriptional repressor ICER and cAMP-induced programmed cell death. Oncogene 15: 827–836
Martens A, Van Bekkum DW and Hagenbeek A . 1990 The BN acute myelocytic leukemia (BNML) (a rat model for studying human acute myelocytic leukemia (AML)). Leukemia 4: 241–257
Hawley RG, Lieu FH, Fong AZ and Hawley TS . 1994 Versatile retroviral vectors for potential use in gene therapy. Gene Ther. 1: 136–138
Pear WS, Nolan GP, Scott ML and Baltimore D . 1993 Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U.S.A. 90: 8392–8396
Duprez E, Gjertsen BT, Bernard O, Lanotte M and Doskeland SO . 1993 Antiapoptotic effect of heterozygously expressed mutant RI (Ala336→Asp) subunit of cAMP kinase I in a rat leukemia cell line. J. Biol. Chem. 268: 8332–8340
Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P and Traganos F . 1992 Features of apoptotic cells measured by flow cytometry. Cytometry 13: 795–808
Jagels MA and Hugli TE . 1994 Mechanisms and mediators of neutrophilic leucocytosis. Immunopharmacology 28: 1–18
Savill J and Haslett C . 1995 Granulocyte clearance by apoptosis in the resolution of inflammation. Semin. Cell Biol. 6: 385–393
Coffey RG . 1992 Effects of cyclic nucleotides on granulocytes. Immunol. Ser. 57: 301–338
Ongini E and Fredholm BB . 1996 Pharmacology of adenosine A2A receptors. Trends Pharmacol. Sci. 17: 364–372
Elferink J and VanUffelen BE . 1996 The role of cyclic nucleotides in neutrophil migration. Gen. Pharmacol. 27: 387–393
Sigurdsson F, Khanna-Gupta A, Lawson N and Berliner N . 1997 Control of late neutrophil-specific gene expression: insights into regulation of myeloid differentiation. Semin. Hematol. 34: 303–310
Borner C . 1996 Diminished cell proliferation associated with the death-protective activity of Bcl-2. J. Biol. Chem. 271: 12695–12698
Vairo G, Innes KM and Adams JM . 1996 Bcl-2 has a cell cycle inhibitory function separable from its enhancement of cell survival. Oncogene 13: 1511–1519
Park J, Robertson K, Hickstein DD, Tsai S, Hockenbery DM and Collins SJ . 1994 Dysregulated bcl-2 expression inhibits apoptosis but not differentiation of retinoic acid-induced HL-60 granulocytes. Blood 84: 440–445
Lagasse E and Weissman IL . 1994 Bcl-2 inhibits Apoptosis of neutrophils but not their engulfment by macrophages. J. Exp. Med. 179: 1047–1052
Fairbairn LJ, Cowling GJ, Reipert BM and Dexter TM . 1993 Suppression of apoptosis allows differentiation and development of a multipotent hemopoietic cell line in the absence of added growth factors. Cell 74: 823–832
Maniatis T, Fritsch EF and Sambrook J . 1989 Molecular Cloning, A Laboratory Manual. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY.
Hillion J, Le Coniat M, Jonveaux P, Berger R and Bernard OA . 1997 AF6q21, a novel partner of the MLL gene in t(6;11)(q21;q23), defines a forkhead transcriptional factor subfamily. Blood 90: 3714–3719
Chomczynski P and Sacchi N . 1987 Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162: 156–159
Williams G . 1987 High-efficiency preparation of DNA from limiting quantities of eukaryotic cells for hybridization analysis. Gene 53: 121–126
Wright S, Schellenberger U, Wang H, Wang Y and Kinder D . 1998 Chemotherapeutic drug activation of the AP24 protease in apoptosis: requirement for caspase-3-like-proteases. Biochem. Biophys. Res. Commun. 245: 797–803
Acknowledgements
P Séité and S Ruchaud have contributed equally to the achievement of this work. Authors wish to thank Dr. T McDonnell (MD Anderson Cancer Institute, Houston, TX) for providing us with human Bcl-2 cDNA. S Ruchaud was supported by a grant from the Ligue Nationale contre le Cancer. This work has benefited from EU grants (TMR program), Ligue Nationale contre le Cancer, Ligue contre le Cancer des Yvelines, Fondation de France, Association pour la Recherche contre le Cancer and the Norwegian Research Council.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by J Savill
Rights and permissions
About this article
Cite this article
Séité, P., Ruchaud, S., Hillion, J. et al. Ectopic expression of Bcl-2 switches over nuclear signalling for cAMP-induced apoptosis to granulocytic differentiation. Cell Death Differ 7, 1081–1089 (2000). https://doi.org/10.1038/sj.cdd.4400730
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4400730
Keywords
This article is cited by
-
Anti-microbial and cytotoxic 1,6-dihydroxyphenazine-5,10-dioxide (iodinin) produced by Streptosporangium sp. DSM 45942 isolated from the fjord sediment
Applied Microbiology and Biotechnology (2014)
-
Cyclic AMP induces IPC leukemia cell apoptosis via CRE-and CDK-dependent Bim transcription
Cell Death & Disease (2011)
-
Camptothecin and khat (Catha edulis Forsk.) induced distinct cell death phenotypes involving modulation of c-FLIPL, Mcl-1, procaspase-8 and mitochondrial function in acute myeloid leukemia cell lines
Molecular Cancer (2009)
-
Gap junctions and the propagation of cell survival and cell death signals
Apoptosis (2005)
-
Differentiation, Apoptosis, and Function of Human Immature and Mature Myeloid Cells: Intracellular Signaling Mechanism
International Journal of Hematology (2001)
