
Abstract
Cytochrome c (cyto c) release from mitochondria is a critical event in apoptosis. By investigating the ordering of molecular events during genotoxic stress-induced apoptosis, we found that ionizing radiation (IR) and etoposide induced the release of cyto c from mitochondria in two distinct stages. The early release of low levels of cyto c into the cytosol preceded the activation of caspase 9 and 3, but had no effect on ATP levels or mitochrondrial transmembrane potential (Δψm). In contrast, the late stage cyto c release resulted in a drastic loss of mitochondrial cyto c and was associated with reduction of ATP levels and Δψm. Moreover, caspases contributed to the late cyto c release since the caspase inhibitor zVAD prevented only the late but not the early-stage cyto c release. Recombinant caspase 3 induced cyto c release from isolated mitochondria in the absence of cytosolic factors. Bcl-2 but not Bid was cleaved during apoptosis after caspase activation. This suggests that Bcl-2 cleavage might contribute to the late cyto c release, which results in mitochondrial dysfunction manifested by the decrease of ATP and Δψm. zVAD prevented the reduction of ATP, Δψm, and nuclear condensation when added up to 8 h after IR, at the time the caspases were highly activated but when the majority of cyto c was still maintained in the mitochondria. These findings link the feedback loop control of caspase-induced cyto c release with mitochondrial dysfunction manifested by ATP and Δψm decline.
Similar content being viewed by others


Introduction
The cellular response to ionizing radiation (IR) and other genotoxic agents includes cell cycle arrest and activation of DNA repair.1 When cells are unable to repair the DNA damage inflicted upon treatment with these agents, they respond by initiation of apoptosis, a process consisting of induction (pre-commitment), commitment, and execution (post-commitment) phases during which the cells are eventually degraded. The molecular events underlying this process are best understood for apoptosis induced by Fas or TNF receptor activation, however, the events following genotoxic stress are unclear.
It has been suggested that a critical early event leading to apoptosis is the release of cytochrome c (cyto c) from mitochondria into cytosol.2,3,4,5 Once released into the cytosol, cyto c binds to Apaf-1,6 the human homologue of the C. Elegans Ced-4, and in the presence of dATP or ATP2 forms a complex with caspase-9,6,7 a regulatory member of a family of cysteine proteases, now called caspases.8 This complex then initiates activation of other effector caspases, which cleave cellular proteins at a specific tetrapeptide sequence after aspartate residues.9,10,11 Mitochondria plays an essential role in apoptosis through the redistribution of intermembrane mitochondrial proteins, the best studied of which is cyto c.12 The mechanism of cyto c release from mitochondria during the course of apoptosis is, however, not clear.12 The mitochondrial membrane potential (Δψm), considered to result following the opening of the permeability transition pore, has been reported to be an irreversible step toward apoptosis.12 Bcl-2, a prototypic anti-apoptotic protein, has a predominantly outer mitochondrial membrane location,13 and blocks the reduction in Δψm, and cyto c release.3,4
Agents which induce genotoxic stress, such as IR and chemotherapy, are the major clinical tools for cancer treatment. These treatments induce apoptosis, however, the ordering of the apoptotic molecular events remains unclear. Here we show a different cyto c release during IR-induced apoptosis of myeloid cells, consisting of an early, low level release preceding caspase activation and a late, massive release following it. Our data suggest that caspases, once activated, contribute to amplification of cyto c release, providing a functional link between caspase-induced cyto c release and mitochondrial dysfunction manifested by the decrease in Δψm and ATP.
Results
Genotoxic agents induce caspase activation which is preventable by Bcl-2
Treatment with IR efficiently kills hematopoietic cells,14 including the IM-9 multiple myeloma cells. Annexin V was used to determine phosphatidylserine exposure on the cell membrane in combination with propidium iodide (PI) to establish the integrity of the cell membrane. Membrane changes characteristic of apoptotic cells could be detected by 4 h, with 25%, or 37% of cells becoming Annexin positive by 16 and 24 h, respectively (of which 10%, or 17% of cells being PI-negative). Phosphatidylserine exposure on the cell membrane was effectively prevented by stable Bcl-2 overexpression in IM-9 cells.
Caspase activation is essential in cells induced into apoptosis by many agents including genototoxic stress.14 We examined the status of caspase 9, a regulator caspase protein which can be activated following IR.14 Western analyses showed that the 45 kD caspase 9 protein was partially converted to a 37 kD form as early as 4 h after IR in IM-9 cells (Figure 1A), indicating that caspase 9 was activated at this time. Both the pancaspase inhibitor zVAD and Bcl-2 effectively prevented the appearance of the 37 kD caspase 9 proteolytic fragment. We also examined the activation of effector caspases by determining cleavage of PARP, and that of the chromogenic caspase tetrapeptide substrate Ac-DEVD-pNA, reflecting the intracellular caspase 3 and 7 activity. An 85 kD PARP fragment and DEVD-pNA cleavage activity (2.5 U±0.25 S.E.) appeared by 4 h following IR. The DEVD cleavage activity was further elevated to 6.6 U±0.95 S.E., 8.3 U±0.39, and 7.3 U±0.34 S.E. at 8, 16 and 24 h, respectively, indicating that it was maintained at high levels during the period in which cells underwent apoptosis. Similar results were obtained after treatment with 10 μM etoposide, a widely used anticancer agent, indicating that multiple genotoxic agents induced caspase activation leading to apoptosis (data not shown).

Two distinct stages of cyto c release before and after caspase activation following IR. IM-9 or IM-9/Bcl-2 cells were lysed at the indicated times following IR and the cell lysates were examined for caspase 9 cleavage by immunoblotting with α-caspase 9 polyclonal Antibody. (A) Cyto c levels were determined in the cellular fractions isolated by differential centrifugation as described in ‘Materials and Methods’ from control or irradiated IM-9 or IM-9.Bcl-2 cells. Immunoblotting with α-cyto c mAb was used to determine the levels of cyto c in the S100 (B) and P10 (C) fractions, representing the cytosolic and mitochondrial cyco c, respectively, in 20 μg of cellular protein. Porin and β-actin were used as loading controls
Distinct stages of cyto c release during apoptosis
Previous studies have shown that translocation of cyto c into the cytosol occurs in response to multiple apoptotic stimuli and plays an important role in inducing apoptosis.2,3,4,5 Release of cyto c from mitochondria is a potential signaling component for regulating apoptosis in many cell types including myeloma cells induced into apoptosis by IR.15 Therefore we wished to examine next the redistribution of cyto c in cytosolic and mitochondrial cellular fractions prepared from Dounce-homogenized IM-9 cell lysates following exposure to IR under conditions which kept mitochondria intact. Cyto c was undetectable in cytosol isolated from untreated cells. In contrast, cytosols from irradiated cells contained increased cyto c levels starting at 2 h, as detected by Western blotting. Cyto c levels increased moderately at 4 and 8 h, and more significantly at 16 h after IR (Figure 1B). Cytosolic cyto c levels declined 24 h after IR, possibly due to protein degradation. In fact, immunoblot analyses of whole cell lysates showed that the overall levels of cyto c decreased as well at 24 h (data not shown).
Examination of cyto c levels contained in the mitochondrial P10 cellular fraction revealed only a slight reduction in cyto c levels up to 8 h following IR. In contrast, there was a dramatic loss of cyto c at 16 and 24 h following IR, indicating a depletion of cyto c from mitochondria. Cyto c release was prevented in cells overexpressing Bcl-2 (Figure 1C). These data suggest that there are two stages of cyto c release corresponding to early and late apoptosis, at a time preceding or following caspase activation, respectively. The distinct stages of cyto c release were not limited to IR, since etoposide also induced cyto c release from mitochondria. However, the kinetics of cyto c release following etoposide treatment was faster, the depletion of mitochondrial cyto c being observed by 8 h following VP16 treatment (data not shown).
The late stage cyto c release is associated with a decrease in ATP levels and Δψm
To address the functional significance of the two-stage cyto c release, we examined the physiological relevance of the late stage cyto c release, which results in depletion of mitochondrial cyto c levels. We examined the levels of ATP and the mitochondrial transmembrane potential (Δψm), two key indicators of mitochondrial function. There was no change in the levels of ATP up to 8 h following IR, even though a low level of cyto c was released and caspases were activated. In contrast, there was a substantial decline of ATP levels starting 16 h after IR (Figure 2), a time corresponding to the late stage cyto c release. Importantly, our data suggested that the early maintenance of constant ATP levels corresponded closely to the steady levels of mitochondrial cyto c during the early stages of apoptosis. We next examined changes in Δψm during apoptosis. We found that the intensity of staining with the mitochondria-specific dye Rhodamine 123, known to reflect Δψm,16 was maintained largely up to 8 h following IR. However, there was a significant increase in Rhodamine 123 staining at 16 to 24 h, reflecting a reduction in Δψm (Figure 2), at a time coinciding with a dramatic increase in the release of cyto c from mitochondria. Furthermore, the changes in ATP levels and of Δψm were also blocked by Bcl-2 overexpression. These data indicate that the ATP and Δψm changes are dependent on cyto c depletion from mitochondria and are associated with mitochondrial dysfunction as a result of cyto c depletion, events which can all be prevented by Bcl-2. These data suggest that Δψm and ATP changes could be associated with the second but not the first phase cyto c release.

Decrease of ATP and Δψm are late events in IR-induced apoptosis. Total cellular ATP content was assayed in IM-9 (filled symbols) and IM-9/Bcl-2 (open symbols) cells, by the luciferin/luciferase assay (Sigma), as described.24 Equal numbers of cells were lysed by boiling in 500 μl H2O and 50 μl was taken for ATP measurements (○) with a luminometer (Biometra). The data were normalized against the control and represented as the mean value of three separate experiments. Δψm (▪) was determined on 4×105 cells washed in PBS following IR, and incubated with 0.5 μM Rhodamine 123 for 30 min at 37°C before analysis by flow cytometry. The data were normalized against that of untreated cells, with values representing means±s.d. (n=4; two separate duplicate experiments). The specificity of the Δψm measurements was confirmed using carbonyl cyanide m-chlorophenylhydrazone (mCCCP), an uncoupling agent that abolishes Δψm. Moreover, the change in Δψm was not due to a change of mitochondrial mass, since there was no difference between control or irradiated cells (up to 24 h) in staining with nonyl acridine orange (NAO, a typical marker for mitochondrial mass)
Caspases induce late cyto c release and Bcl-2 cleavage
To address whether caspase activation is associated with the late stage cyto c release, we examined cyto c release in cells in culture following IR in the absence, or presence, of zVAD. zVAD but not the caspase 1 inhibitor YVAD, effectively prevented DEVD cleavage activity when assayed 4 or 18 h following IR (data not shown). zVAD also prevented short-term cytotoxicity, since 24 h following IR there was a loss of viability of 33.19% (±5.41), 1.58% (±0.57), and 24.52% (±2.36) in IR, IR+zVAD, or IR+YVAD-treated cells, respectively. zVAD also blocked specifically the late-stage increase in cytosolic cyto c levels (Figure 3A, left panel). In contrast, zVAD had no effect on cyto c release up to 4 h following IR. The effect of zVAD was not limited to IR-induced cyto c release, since it could also prevent cyto c release during 18 h treatment with VP16 (Figure 3A right panel). These results indicate a potential role for a caspase in the amplification of cyto c release by causing a late, but not early cyto c release. These data suggest that different mechanisms are responsible for the early and late cyto c release.

Caspases induce late cyto c release from mitochondria and cleavage of Bcl-2. (A) Cells were treated with IR or VP16 in the absence or presence of 100 μM zVAD, lysed and the levels of cyto c in the cytoplasmic S100 fractions determined by immunoblotting with α-cyto c mAb, and β-actin as a loading control. (B) The P10 mitochondrial fractions were incubated with recombinant caspase 3 in the absence, or presence, of zVAD at 30°C for 2.5 h. The reactions were then pelleted and the cytosolic S10-2 from IM-9 or IM-9/Bcl-2 cells were examined for cyto c levels. Last lane in bottom panel represents a P10 fraction used as a positive control for cyto c. The mitochondrial protein MTCO1 was not detected in S10-2 fractions. (C) Bcl-2 expression was determined at the indicated times following IR in the absence or presence of 100 μM DEVD. (D) S100 from apoptotic (24 h after IR) IM-9 cells were incubated with P10 from untreated cells in the absence or presence of 100 nM zVAD or YVAD to examine in vitro cleavage of Bcl-2. Bcl-2 was analyzed by Western blotting using an α-Bcl-2 mAb. (E) Whole cell lysates of IM-9 cells were examined for expression of Bid following different times after IR by Western blotting using an α-Bid polyclonal antibody.18 The data shown are representative of three different experiments
To directly examine the role of caspase 3 in cyto c release, we next used an in vitro reconstituted system consisting of mitochondrial fractions and recombinant caspase 3. Examination of the supernatant of this reaction (post-mitochondrial fraction, designated S10-2) revealed a dose-dependent increase in mitochondrial cyto c content (Figure 3B). Cyto c was not detected in S10-2 isolated from untreated P10 fractions, and it could be prevented in the presence of 2 or 20 μM zVAD. Moreover, in vitro cyto c release could be effectively prevented by Bcl-2 since S10-2 isolated from Bcl-2 overexpressing cells did not contain cyto c when treated with caspase 3 under identical conditions (Figure 3B). In addition, examination of the caspase 3-treated mitochondrial P10-2 cellular fraction revealed a dose-dependent decrease in mitochondrial cyto c content, which was prevented by zVAD (data not shown). These data indicate that caspase 3 directly contributes to the late cyto c release in the absence of cytosolic factors.
Next we sought to identify the mitochondrial target(s) of caspase-3. Immunoblotting of IR-treated IM-9 cells with anti-Bcl-2 mAbs revealed the appearance of a novel, 23-kD band (Bcl-2Δ), in addition to the expected 26-kD band corresponding to the Bcl-2 protein. This novel Bcl-2 protein species appeared starting 8 h following 10 Gy IR and increased in intensity to 30 h and was inhibited by DEVD (Figure 3C). We further examined Bcl-2 cleavage in vitro, when P10 from IM-9 cells containing endogenous Bcl-2 was incubated with S100 isolated from IM-9 cells 24 h after IR. There was an abundant production of the p23 kD fragment, which was prevented by 100 nM zVAD but not YVAD (Figure 3D). The p23 Bcl-2Δ fragment generated in this assay, or when in vitro translated recombinant Bcl-2 was incubated with purified recombinant caspase-3 (Q Chen and A Almasan, unpublished), is consistent with cleavage at a conserved aspartic acid residue at position 34, following the caspase 3 consensus tetrapeptide at residues 31-34 (DAGD), as previously reported.17 In contrast, immunoblotting showed that the previously reported p13 and p15 Bid cleavage products associated with cyto c amplification during Fas-induced apoptosis18 were not detected in whole cell lysates from apoptotic IM-9 cells and the levels of Bid did not change (Figure 3E), indicating that Bid could not be responsible for the cyto c release in our system.
zVAD prevents mitochondrial dysfunction and nuclear fragmentation
We next examined the association of caspase activation with mitochondrial dysfunction and nuclear morphological changes. Since caspase targeting to mitochondria resulted in cyto c release, we examined the effect of zVAD on ATP and Δψm, when zVAD was added at different times following IR. zVAD blocked effectively any significant reduction of both ATP and Δψm, even when it was added to cells 8 h after IR (Figure 4A). In contrast, zVAD had no effect when it was added 16 h after IR, a time corresponding to the second stage cyto c release.

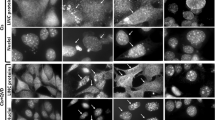
zVAD blocks ATP and Δψm reduction, as well as nuclear fragmentation. (A) IM-9 cells were cultured in the absence or presence of 75 μM zVAD, added at the indicated times after IR. Δψm (closed bars) and ATP (open bars), was examined at 24 h after IR, as described for Figure 2. (B) nuclear condensation was examined at 24 h after IR by Hoechst 33258 staining and fluorescent microscopy in irradiated IM-9 cells. At least 200 cells were examined; values represent means±S.D. (n=4; two separate duplicate experiments). (C) Cells were immunostained with either cyto-c specific mAbs or the mitochondria-specific dye, MitoTracker Red. The α-cyto c mAbs were detected with FITC-conjugated α-mouse secondary antibodies. The nuclei were stained by ToPRO-3. On the right is an overlay of the three images. Magnification is ×400
We next examined the nuclear morphological changes associated with IR-induced apoptosis. Staining with the DNA-intercalating dye Hoechst 33258 revealed a significant number of cells with nuclear condensation characteristic of apoptotic cells 24 h after IR (Figure 4B). Addition of zVAD up to 8 h following IR effectively prevented nuclear condensation 24 h after IR, but it had no effect when it was added 16 h following IR. To further monitor the consequences of mitochondrial cyto c loss in individual cells, we used confocal microscopy. Following 0 (top) or 8 h (data not shown) after IR, cells displayed a punctate cyto c staining (Figure 4C). Cyto c co-localized with the mitochondria-specific dye, MitoTracker Red. Following 24 h after IR, cyto c staining became diffuse, indicating cyto c loss from mitochondria and its translocation to the cytosol. No cells were observed with nuclear fragmentation, as determined by ToPRO-3 staining, in the absence of diffuse cyto c staining. These indicate that the late, massive cyto c release preceded the appearance of nuclear fragmentation.
Discussion
In an effort to elucidate the ordering of molecular and cellular events in IR-induced apoptosis, we found that cyto c was released in two distinct stages, with caspase activation being required for the late stage massive cyto c release, resulting in depletion of cyto c from mitochondria and associated mitochondrial dysfunction. We clearly demonstrate that the initial cyto c release is characterized by release of only a small fraction of mitochondrial cyto c. This is apparently sufficient to trigger caspase 9 and 3 activation in the multiple myeloma IM-9 cells in response to the genotoxic stress caused by γ-irradiation and etoposide. The low levels of cyto c released early into the cytosol did not significantly impact on the remaining mitochondrial cyto c levels or mitochondrial activity as measured by ATP levels or changes in Δψm. Our findings reconcile the dual role of cyto c for an adequate oxidative phosphorylation necessary for ATP production,12,19 and as a source for caspase activation in the cytosol, with an expected loss of ATP following mitochondrial cyto c depletion.
The late cyto c release can be a consequence of activated caspases since recombinant caspase 3 can cause cyto c release from mitochondria and zVAD blocks cyto c release from isolated mitochondria and late-stage apoptotic cells, as well as the ATP and Δψm drop associated with mitochondrial cyto c depletion. Consistent with our data, it has been recently shown that several caspases, including caspase 3, can also induce permeability transition pore opening in a reconstituted system.20 Recently, it has been reported that caspases can induce cyto c release in a cell free system in the presence of a cytosolic factor.21 However, our data suggest that the caspase 3-induced cyto c release might not require cytosolic factors. At least in myeloid cells undergoing IR-induced apoptosis Bid does not seem to be required for this late cyto c release since the Bid cleavage products were not detected using an antibody identical to the one used in previous reports.18 Rather, we found Bcl-2 to be a caspase 3 target, although additional mitochondrial caspase targets, yet to be identified, might be also involved. Bcl-2 cleavage is likely to contribute to cyto c release, since, once it is truncated, it becomes a pro-apoptotic Bax-like molecule and can induce cyto c release in the presence of zVAD (Q Chen and A Almasan, unpublished).
We also found that targeting of mitochondria by caspases was associated both with mitochondrial dysfunction and nuclear condensation. Intracellular ATP and Δψm reduction apparently occurred after caspase activation and were inhibitable by zVAD and Bcl-2. Interestingly, Δψm reduction and nuclear condensation could be prevented even when zVAD was added 8 h following IR, at a time when caspases were highly activated, but the majority of cyto c was maintained in the mitochondria, suggesting that the caspase-induced cyto c release is a necessary step for the post-commitment phase of apoptosis. These cells may commit to undergo apoptosis before the initial cyto c release since zVAD and Bcl-2 can not provide a clonogenic survival advantage to IM-9 cells (data not shown). In non-genotoxic stress-induced apoptosis, halting cyto c release may also prevent apoptosis. Thus, in case of the v-Abl oncoprotein-suppressed apoptosis induced by withdrawal of IL-3, providing cells with survival factors stopped further cyto c release and conferred clonogenic survival to the remaining cell population.5
It is possible that, as a result of caspase targeting of mitochondria, cells may switch from apoptosis to secondary necrosis, a phenomenon associated with late apoptosis. Perhaps, as reported, ATP could be the determining factor for the cellular decision to undergo apoptosis or necrosis.19 Indeed, ATP is required for apoptosis, with ATP and dATP being reported to be required cofactors for cyto c-induced caspase 9 activation by the cyto c/Apaf-1 complex,2 and the presence of ATP has been shown to play a critical role in determining the mode of cell death, apoptosis vs necrosis.19 As in our study, ATP levels were also reported to be unchanged during the early stages of apoptosis of UV-B and staurosporine-treated T lymphoblastoid CEM cells.22 To maintain the mitochondrial cyto c needed for sustained ATP production, it is possible that only the excess cyto c is initially released.12 The mechanism of the initial cyto c release following genotoxic stress is currently under investigation.
The molecular events leading to apoptosis are best characterized for Fas-induced cell death. Although the engagement of Fas by Fas ligand can directly activate caspase-8 and its downstream cascade, a late stage amplification of cyto c release by caspases is likely to take place following caspase 8-mediated cleavage of Bid, a pro-apoptotic Bax homologue.18 However, Bid cleavage might not be responsible for cyto c release following IR or etoposide treatment of IM-9 cells, since Bid cleavage was not detected, nor were any changes in Bid levels or in its subcellular localization. It was suggested that the caspase-associated cyto c release happened in the Fas- but not etoposide-induced apoptosis since zVAD could not inhibit etoposide induced cyto c release.23 Thus, to our knowledge, this is the first report showing that IR and etoposide-induced caspase activation can contribute to the late cyto c release. Importantly, our results suggest that zVAD can inhibit the late cyto c release but not the early one induced by IR. Clearly, both the genotoxic stress and the Fas-induced apoptotic pathways seem to share a somewhat similar late-stage caspase amplification following mitochondrial targeting by caspases, since ultimately caspase 3 is activated in both systems. Whether caspase-9 activated by IR contributes to cyto c release as suggested21 and whether another mitochondrial protein is an additional caspase target needs to be further explored.
In summary, we present evidence for a differential cyto c release during early and late genotoxic stress-induced apoptosis. We show that there is a positive feedback amplification of cyto c release by caspases and identify Bcl-2 as a caspase target, which may contribute to the late cyto c release. Importantly, our data indicate that cyto c amplification is associated with mitochondrial dysfunction manifested by ATP and Δψm reduction, which may further lead to nuclear condensation. These data suggest that the feedback loop control of late cyto c release by caspases is an important step required for the post-commitment phase of apoptosis, necessary because cells must utilize diverse mechanisms, even after caspase activation, to assure an ordered progression toward death.
Materials and Methods
Cell culture and transfections
Human IM-9 multiple myeloma cells were obtained from ATCC and cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Gibco). Exponentially growing cells were subjected to 10 Gy γ-irradiation from a 137Cs source, as described,24 or cultured in media containing 10 μM etoposide. pSFFV-neo FLAG.Bcl-2 was used for stable transfection of IM-9 cells. Cell clones overexpressing Bcl-2 were selected in 1 mg/ml G418. One representative clone, IM-9/Bcl2.cl1-1, which abundantly expressed FLAG.Bcl-2 was further used.
Protease activity assays
Cells (3×105) were lysed on ice and caspase assays performed as described.14 Substrate only was used for background control and the Ac-DEVD-pNA cleavage activity was normalized against untreated cells. His-tagged PET-CPP, encoding recombinant caspase-3 [a gift from Drs. ES Alnemri and SM Srinivasula] was transformed into E. coli BL21 and purified by His6-resin affinity purification (Novagen). The freshly prepared caspase 3 protein (1 U=10 pmol/min/mg) was examined for its ability to cleave the DEVD-pNA substrate before being used for further experiments. Recombinant caspase-3 was mixed with 5 μl P10 prepared from parental IM-9 and derivative cells stably expressing Flag-Bcl-2 (about 1 mg protein/ml) in a total of 40 μl of buffer A, and incubated for 2.5 h at 30°C. For certain experiments, purified caspase-3 was first incubated with zVAD for 15 min at room temperature. These P10-containing reactions were then pelleted at 10 000×g for 8 min and the resulting supernatant (S10-2) and pellet (P10-2) were then subjected to SDS–PAGE and immunoblotting for cyto c and Bcl-2 as described above. Caspase peptide inhibitors or substrates were from Calbiochem; all other chemicals were from Sigma, unless otherwise specified.
Cell fractionation and immunoblot analyses
Cells were fractionated as previously reported.2,3,5,16 Briefly, cells were homogenized with a Dounce homogenizer (5–8 strokes) in buffer A containing 20 mM HEPES-KOH (pH 7.2), 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 250 mM sucrose, and protease inhibitors. The homogenate was centrifuged at 750×g for 5 min and the supernatant further centrifuged at 10 000×g. This mitochondria-containing pellet was designated P10 and the supernatant was subjected to further ultracentrifugation at 100 000×g for 45 min. The resulting pellet and supernatant, representing the ER and cytosolic fractions, were designated as P100 and S100, respectively. The purity of the cellular fractions was examined by Western blotting using α-calnexin, α-PCNA, and α-MTCO1 mAbs, as ER, nuclear and mitochondrial markers. Bcl-2 was contained mainly in P10, DEVD cleavage activity in S100, with cyto c being absent from the P100 fractions.
For immunoblots, cells and membrane fractions were washed in PBS and lysed in 1% NP-40 buffer. Cellular proteins were separated by SDS–PAGE, transferred onto nitrocellulose membranes, probed using immunoblot analyses with antibodies specific to the human β-actin (Sigma), Bcl-2, caspase 9, PCNA (Santa Cruz), Calnexin (Transduction Laboratories), cyto c (6581A for Western and 65971A for immunostaining; Pharmingen), cyto c oxidase I (MTCO1; Molecular Probes), or porin (Calbiochem), followed by incubation with a secondary antibody conjugated to horseradish peroxidase for 1 h at room temperature. Antibodies to Bid18 were a generous gift from Dr. X Wang (U.T. Southwestern). Immunoreactive bands were detected using enhanced chemiluminescence (Amersham) and visualized by autoradiography.
Flow cytometric and immunofluorescence analyses
Phosphatidylserine exposure on cell membranes was determined as described.14 Δψm was determined following incubation with 0.5 μM Rhodamine 123 for 30 min at 37°c before analysis by flow cytometry. The data were analyzed by Cell-Quest and the mean value of FL-1 (530 mn) were collected after the non-viable cells were gated out. The data were then normalized against that of untreated cells and evaluated in reference to: (i) 5 μM m-chlorophenylhydrazone, an uncoupling agent that abolishes Δψm, or (ii) 100 nM nonyl acridine orange, a mitochondrial mass indicator.24
For cyto c subcellular localization cells were grown on Vitrogen-coated coverslips, washed with PBS, fixed with 2% paraformaldehyde, incubated with α-cyto c mAb, followed by FITC-conjugated secondary mAb. MitoTracker Red (100 nM) was used for mitochondrial localization, ToPRO-3 (15 nM) and Hoechst 33258 (10 μg/ml) for nuclear staining in confocal and conventional microscopy, respectively. The immunostained slides were examined using a Nikon FXA epifluorescence microscope or a Leica TCS-NT confocal laser scanning microscope with an 63×objective lens (Leica Lasertechnik GmbH, Heidelberg, Germany). Confocal images were examined along the z-axis with a step-size of 1 μm. For comparison of triple-stained patterns the three images were superimposed. MitoTracker Red, ToPRO-3, and Hoechst 33258 were from Molecular Probes.
Abbreviations
- cyto c:
-
cytochrome c
- DEVD:
-
acetyl-Asp-Glu-Val-Asp
- fmk:
-
fluorometyl ketone
- mAb:
-
monoclonal antibody
- pNA:
-
p-nitroanilide
- YVAD:
-
acetyl-Asp-Glu-Val-CHO
- zVAD:
-
benyloxycarbonyl-Val-Ala-Asp-fmk
- Δψm:
-
mitochondrial membrane potential
References
Giaccia AJ and Kastan MB . (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev. 12: 2973–2983
Liu X, Kim CN, Yang J, Jemmerson R and Wang X . (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86: 147–157
Kluck R, Bossy-Wetzel E, Green D and Newmeyer D . (1997) The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 275: 1132–1136
Yang J, Liu X, Bhalla K, Kim C, Ibrado A-M, Cai J, Peng T-I, Jones DP and Wang X . (1997) Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science 275: 1129–1132
Chen Q, Takeyama N, Brady G, Watson AJM and Dive C . (1998) Blood cells with reduced mitochondrial membrane potential and cytosolic cytochrome c can survive and maintain clonogenicity given appropriate signals to suppress apoptosis. Blood 92: 4545–4553
Zou H, Henzel WJ, Liu X, Lutschg A and Wang X . (1997) Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90: 405–413
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES and Wang X . (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479–489
Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW and Yuan J . (1996) Human ICE/CED-3 protease nomenclature. Cell 87: 171
Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin TT, Yu VL and Miller DK . (1995) Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 376: 37–43
Fernandes-Alnemri T, Litwack G and Alnemri ES . (1995) Mch2, a new member of the apoptotic Ced-3/Ice cysteine protease gene family. Cancer Res. 55: 2737–2742
Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS and Dixit VM . (1995) Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81: 801–809
Green DR and Reed JC . (1998) Mitochondria and apoptosis. Science 281: 1309–1312
Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W and Reed JC . (1993) Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 53: 4701–4714
Gong B, Chen Q, Endlich B, Mazumder S and Almasan A . (1999) Ionizing radiation-induced, Bax-mediated cell death is dependent on activation of multiple serine and cysteine proteases. Cell Growth Differ. 10: 491–502
Chauhan D, Pandey P, Ogata A, Teoh G, Krett N, Halgren R, Rosen S, Kufe D, Kharbanda S and Anderson K . (1997) Cytochrome c-dependent andindependent induction of apoptosis in multiple myeloma cells. J. Biol. Chem. 272: 29995–29997
Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT and Thompson CB . (1997) Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell 91: 627–637
Cheng EHY, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K and Hardwick JM . (1997) Conversion of Bcl-2 to a Bax-like Death Effector by Caspases. Science 278: 1966–1968
Luo X, Budihardjo I, Zou H, Slaughter C and Wang X . (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94: 481–490
Eguchi Y, Shimizu S and Tsujimoto Y . (1997) Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 57: 1835–1840
Marzo I, Susin SA, Petit PX, Ravagnan L, Brenner C, Larochette N, Zamzani N and Kroemer G . (1998) Caspases disrupt mitochondrial membrane barrier function. FEBS Lett. 427: 198–202
Bossy-Wetzel E and Green DR . (1999) Caspases induce cytochrome c release from mitochondria by activating cytosolic factors. J. Biol. Chem. 274: 17484–17490
Bossy-Wetzel E, Newmeyer DD and Green DR . (1998) Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 17: 37–49
Sun XM, MacFarlane M, Zhuang J, Wolf BB, Green DR and Cohen GM . (1999) Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J. Biol. Chem. 274: 5053–5060
Gong B, Chen Q and Almasan A . (1998) Ionizing radiation stimulates mitochondrial gene expression and activity. Radiat. Res. 150: 505–512
Acknowledgements
We would like to thank Drs. SM Srinivasula, ES Alnemri (Thomas Jefferson University) for the PET-CPP, and G Nunez (University of Michigan) for the pSFFV-FLAG.Bcl-2 constructs, X Wang (U.T. Southwestern) for the Bid antibody, J Drazba for confocal microscopy and Ms. A Raber (Flow Cytometry Core facility of Cleveland Clinic) for flow cytometry; The FACS Vantage Cell Sorter was funded by the W.M. Keck Foundation. We also thank Drs. Chris Campbell and S Mazumder for critical reading of the manuscript. This work was supported in part by research grants from the US National Institutes of Health (RO1-CA81504) and RSNA.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by J Reed
Rights and permissions
About this article
Cite this article
Chen, Q., Gong, B. & Almasan, A. Distinct stages of cytochrome c release from mitochondria: evidence for a feedback amplification loop linking caspase activation to mitochondrial dysfunction in genotoxic stress induced apoptosis. Cell Death Differ 7, 227–233 (2000). https://doi.org/10.1038/sj.cdd.4400629
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4400629
Keywords
This article is cited by
-
Antileukemic effect of venetoclax and hypomethylating agents via caspase-3/GSDME-mediated pyroptosis
Journal of Translational Medicine (2023)
-
Glutathione Peroxidase 3 induced mitochondria-mediated apoptosis via AMPK /ERK1/2 pathway and resisted autophagy-related ferroptosis via AMPK/mTOR pathway in hyperplastic prostate
Journal of Translational Medicine (2023)
-
Selenium Alleviates Ammonia-Induced Splenic Cell Apoptosis and Inflammation by Regulating the Interleukin Family/Death Receptor Axis and Nrf2 Signaling Pathway
Biological Trace Element Research (2023)
-
Chlorogenic acid induces apoptosis and cell-cycle arrest in colorectal cancer cells
Molecular Biology Reports (2023)
-
Targeting BCL-2 in B-cell malignancies and overcoming therapeutic resistance
Cell Death & Disease (2020)
