
Abstract
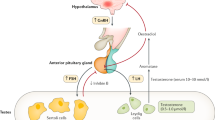
Hypogonadotropic patients may visit pediatricians, general practitioners, endocrinologists or urologists, presenting with microphallus, cryptochidism or pubertas tarda and delayed bone maturation. Congenital hypogonadotropic hypogonadism is characterized, apart from small testes, by the constellation of low serum levels of testosterone, LH and FSH. Kallman's syndrome is characterized by congenital hypogonadotropic hypogonadism with midline defects such as anosmia (a deficiency of the sense of smell). The first case report dates back to 1856, and genetic defects causing the syndrome have been recently described. The diagnosis can be clinically suspected and is established by confirming hormonal studies.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 8 print issues and online access
$259.00 per year
only $32.38 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
John, H., Schmid, C. Kallmann's Syndrome: clues to clinical diagnosis. Int J Impot Res 12, 121–123 (2000). https://doi.org/10.1038/sj.ijir.3900493
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ijir.3900493
