
Abstract
Regional genomic alterations resulting from single-copy allelic loss or gain have been well characterized in many human cancers and are often of prognostic relevance. Unbalanced gain of 17q material is common in malignant human neuroblastomas and typically results from unbalanced translocations. Unbalanced 17q gain may be an independent predictor of disease outcome, but technical difficulties with quantifying such gain using fluorescent in situ hybridization gives this method limited clinical applicability. We now describe a duplex genomic DNA-based quantitative polymerase chain reaction assay to determine the presence or absence of unbalanced gain of chromosome 17q in primary neuroblastoma specimens. The technique was first refined and validated in a panel of nine human neuroblastoma-derived cell lines by direct comparison with dual-color fluorescent in situ hybridization. Prospective blinded comparison of quantitative polymerase chain reaction and fluorescence in situ hybridization in 40 human neuroblastoma primary tumor samples showed a sensitivity of 96% and 100% specificity for detecting unbalanced 17q gain when a relative 17q copy number ratio of 1.3 was used to define unbalanced gain. Tumors with ratios >1.3 were highly associated with malignant tumor phenotypic features such as metastatic disease (P < .0001) and tumor MYCN amplification (P = .008). These data suggest that quantitative polymerase chain reaction determination of 17q status is feasible and highly specific in primary tumor samples. Sensitivity may be limited because of the inherent complexity of both the chromosomal rearrangements and heterogeneity of some tumor samples. Taken together, quantitative polymerase chain reaction can be used as a high-throughput screening tool for 17q aberrations, but a subset of samples may also require fluorescence in situ hybridization analysis in an attempt to conclusively determine 17q allelic status.
Similar content being viewed by others


Main
Neuroblastoma is the most common malignant disease of infancy and the third most common pediatric cancer overall (1). It is a markedly heterogeneous disease, characterized by a wide diversity in natural history and treatment outcomes. Although patient age at diagnosis and stage of the tumor have been demonstrated to be important variables for predicting survival, it is clear that cellular and molecular features of individual primary tumors convey important additional prognostic information (2). For example, the observation that MYCN amplification is associated with advanced stage of disease and rapid tumor progression (3, 4) has resulted in the current standard of care of testing all diagnostic neuroblastoma samples for this genomic aberration to guide treatment planning. The Children’s Oncology Group, as well as most international pediatric oncology cooperative groups, considers age at diagnosis, disease stage, Shimada histopathologic classification, DNA index, and MYCN amplification status in assigning patients a risk category for subsequent treatment planning (1, 5, 6). However, risk classification using these five prognostic variables remains imprecise, and current research is focused on identifying additional tumor-specific prognostic variables that will improve the ability to properly stratify patients for treatment planning.
Unbalanced gain of 17q genomic material is a common somatically acquired abnormality in primary neuroblastomas (7, 8, 9). It has been demonstrated that these aberrations are frequently caused by unbalanced translocations that concomitantly result in loss of distal 1p, 11q, or other regions of recurrent allelic loss (7, 10, 11, 12, 13, 14). Unbalanced 17q gain is present at diagnosis in the more aggressive subset of neuroblastomas, suggesting that this is a somatically acquired event that contributes to a more malignant phenotype. Furthermore, the review by Bown and colleagues of 313 cases from European centers showed that unbalanced 17q gain detected by FISH was an independent marker for disease outcome (9). These data strongly suggest that risk stratification can be refined by reliable assessment of 17q allelic status in primary neuroblastoma specimens.
Dual-colored interphase FISH on tumor touch preparations appears to be a relatively sensitive and specific method for determining 17q allelic status, but this has not been tested formally. Routine FISH detection of 17q anomalies is difficult in the cooperative group setting because of the large volume of samples requiring analysis, low throughput, high cost, inadequate diagnostic material in many cases relative to what is required for high-quality touch preparations, and intratumoral heterogeneity, often making interpretation difficult. The Children’s Oncology Group is interested in testing the independent prognostic impact of 17q allelic status, especially because the previous report did not include Shimada histopathology as a covariate (9), and histopathologic classification is one of the cornerstones of the Children’s Oncology Group risk stratification system (1). We therefore sought to develop a relatively simple and high-throughput technique using real-time quantitative PCR (Q-PCR), and thus requiring nanogram quantities of input DNA, to screen for unbalanced 17q gain in neuroblastoma specimens.
MATERIALS AND METHODS
Samples and DNA Isolation
Nine human neuroblastoma–derived cell lines were selected from the Children’s Hospital of Philadelphia neuroblastoma cell line repository. Primary tissue samples from 40 diagnostic neuroblastoma specimens were randomly selected from the Children’s Hospital of Philadelphia and Children’s Oncology Group Neuroblastoma Tumor Banks. Each primary tumor sample was immediately snap-frozen in liquid nitrogen on receipt in the pathology lab, according to Children’s Oncology Group guidelines. Clinical and biological variables including age at diagnosis, disease stage by the International Neuroblastoma Staging System (15), DNA index (6), and Shimada pathology (5) were available for all cases, and MYCN amplification status was available by FISH (16) in 38/40 cases. Tumor touch preparations were made from each primary tumor and fixed in 3:1 methanol:acetic acid. DNA was extracted by anion exchange chromotography (Qiagen, Valencia, CA). The Children’s Hospital of Philadelphia Institutional Review Board approved this research.
Real-Time Quantitative PCR
Q-PCR amplimers were designed to be complementary to genomic regions that showed gain (17q) or loss (17p) in all previously reported cases of unbalanced 17q gain (9, 11, 17, 18, 19, 20). Oligonucleotide primers and probes were designed with Primer Express v1.5 (Applied Biosystems, Foster City, CA) and were complimentary to the intron 1 genomic sequence of TP53 and the exon1–intron1 boundary within the genomic sequence of MPO (Table 1). The probes were differentially labeled with fluorophores VIC and FAM to allow for duplex PCR reactions, and each probe was dual labeled with TAMRA to serve as a quencher dye.
Duplex Q-PCR was performed using an ABI PRISM 7700 Sequence Detector. Initial amplification mixtures (25 μL) contained 10 ng of genomic DNA, 1 × TaqMan Universal PCR Master Mix (Applied Biosystems), 360 nm of each primer, and 100 nm of each probe. Final oligonucleotide concentrations were determined by optimization experiments in which primer concentrations were varied between 270 nm and 900 nm and probe concentrations were varied between 20 nm and 200 nm to identify combinations yielding optimal signal-to-noise and amplification efficiency. Cycling conditions were 2 minutes at 50° C and an initial denaturation step at 95° C for 10 minutes, followed by 40 cycles of 95° C for 15 seconds and 60° C for 60 seconds.
Initial Q-PCR experiments were performed using both the standard curve method and comparative threshold cycle (ΔΔCT) method (21). Standard curves were constructed using serial dilutions of normal human genomic DNA from 50 ng to 3.125 ng, and relative MPO and TP53 copy numbers were extrapolated from the normal DNA standard curve using the CT value of an individual test sample. Relative 17q copy number by the comparative CT method was determined in both test and normal samples according to the following equation: 2−ΔΔCt = (1+E) −ΔCt sample/(1+E)−ΔCt calibrator, where the efficiency (E) of target and reference gene amplifications are equivalent (see validation below), ΔCTsample is the difference in CT between the target gene and the reference gene in the test sample, and ΔCTcalibrator is the difference in threshold cycle value between the target gene and the reference gene in the normal DNA sample.
Dual-Color Interphase FISH
FISH probes were designed to overlap the Q-PCR amplimer genomic regions. DNA extracted from human bacterial artificial chromosomes clones RP11–89D11, mapping to the TP53 locus at 17p13.1, and RP11–506H21, mapping to the MPO locus at 17q23.1, were identified (Children’s Hospital of Oakland Research Institute, Oakland, CA), and DNA from each clone was labeled with the Vysis Nick Translation Kit (Vysis, Inc., Downers Grove, IL). The TP53 and MPO probes were labeled with Spectrum Red and Spectrum Green dUTP, respectively. Dual-color interphase FISH was performed according to the manufacturer, with minor modifications. Briefly, slides were subject to denaturation at 73° C in a solution of 70% formamide in 2 × standard saline citrate and then dehydrated in ethanol. Bacterial artificial chromosomes probes were precipitated in the presence of COT-1 DNA and then denatured for 5 minutes at 73° C in LSI/WCP Hybridization Buffer (Vysis, Inc., Downers Grove, IL). Ten microliters of each probe was applied concomitantly to tumor touch preparation slides and hybridized in a moist chamber at 37° C overnight. Slides were washed with 0.3% NP-40/0.4 × standard saline citrate at 73° C, and then with 0.1% NP-40/2 × standard saline citrate at room temperature, and finally they were counterstained with DAPI II.
Fluorescence signals were visualized with a Zeiss Axiophot microscope (Jena, Germany), photographed with a computer-driven Photometrics Sensys color camera, and representative images were analyzed using the Powergene system. Hybridization signals from a minimum of 75 nuclei were counted for each sample. Samples were considered to have unbalanced gain of 17q material if >50% of nuclei demonstrated one or more excess 17q signals relative to 17p signals, whereas ≤50% of nuclei with this pattern was considered no gain.
Statistical Analyses
Clinical and biological features were compared using the exact conditional χ2 test.
RESULTS
Optimization and Validation of Q-PCR in Neuroblastoma Cell Lines
The optimal reaction concentrations determined in the serial dilution experiments were identical for both genes at 360 nm for each primer and 100 nm for each probe (data not shown). Duplex Q-PCR was then performed on serial dilutions of normal genomic DNA. PCR reaction kinetics were similarly efficient for each gene (R ≥ 0.99), suggesting that a comparative CT (ΔΔCT) method was an acceptable strategy for relative quantification of 17q copy number (Fig. 1) (21). Both the standard curve and the ΔΔCT methods were used to determine 17q status in nine neuroblastoma cell lines. Results using both methods were essentially identical (Fig. 2; Table 2). The ΔΔCT method was therefore used for further protocol development because it allows for higher sample throughput by obviating the need for serial dilutions in each experiment.

Quantitative PCR standard curves constructed with normal human genomic DNA. The amount of template input DNA is plotted against CT value for both TP53 (dashed) and MPO (solid). All reactions were performed in duplex with both sets of primers and probes. At each serial dilution, the difference between the TP53 and MPO CT values remains constant, indicating that the efficiencies of the reference amplification (TP53) and the target amplification (MPO) are equivalent. This relationship between the primer-probe sets validates the use of the comparative CT method for determining the presence of 17q gain.

Comparison of standard curve and comparative CT methods for detecting 17q status in human neuroblastoma–derived cell lines. For each cell line tested, the ratio of MPO copy number to TP53 copy number is shown using both the comparative CT method (solid) and the standard curve method (open). All cell lines with unbalanced 17q gain by FISH showed relative 17q copy number ratios by Q-PCR of >1.3.
We next compared Q-PCR results with copy number determination by dual color interphase FISH. In our sample set of nine cell lines, NBLS was the only example of a normal 17p-17q allelic ratio by FISH (Fig. 2; Table 2), and this is not surprising because of the high correlation between 17q gain and the high-risk phenotype that is required to establish cell lines. This was the only sample that showed a relative 17q copy number determination of <1.3. The remaining samples showed a variety of copy number patterns by FISH with either a one- or two-copy difference between 17q and 17p. Relative 17q copy numbers ranged from 1.35–3.39, and these values in general correlated with the degree of discrepancy in 17p and 17q allelic copy number by FISH (Table 2).
Prospective Evaluation of Q-PCR in Neuroblastoma Primary Tumors
Q-PCR and dual-color interphase FISH were performed on 40 primary human neuroblastoma samples by individual investigators who were blinded to the results for the alternate technique. All samples were classified as unbalanced 17q gain present or absent by dual-colored FISH and Q-PCR (Fig. 3; Table 3). All samples could be categorized by FISH, although many samples had complex patterns of 17q rearrangements with multiple distinct fluorescence signal patterns. Table 3 shows the predominant pattern observed. Q-PCR results ranged from 0.69–2.84 and correlated with the relative 17q copy number determination by FISH (Fig. 4). We explored different relative 17q copy number values for the binary determination of presence or absence of unbalanced 17q gain (Table 4). Using a relative 17q copy number of 1.4 as a cut point showed 100% specificity (no false positives), but 6 false negatives (74% sensitivity). Relaxing the stringency to 1.3 again showed 100% specificity with only 1 false negative (96% sensitivity). Specificity decreased to 76% when the relative 17q copy number cut point was further relaxed to 1.2, but the sensitivity was 100%.

Correlation of Q-PCR and FISH in two primary tumors. Amplification plots of MPO (A) and TP53 (B) fluorescent signal versus cycle number for primary neuroblastomas 855 and 2054. The amplification plots are essentially identical for TP53, but tumor 2054 has a lower CT value for MPO. Using the ΔΔCT method, these results correspond to a 17q-17p ratio of 1.9 for tumor 2054 and of 1.0 for tumor 855. These data are confirmed by dual-colored interphase FISH using probes to MPO (green) and TP53 (red). Shown are representative single nuclei showing no gain of 17q material for 855 (C) and unbalanced 17q gain for 2054 (D).

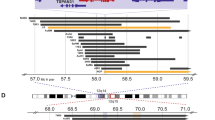
Chromosome 17 status of primary human neuroblastoma specimens, as determined by Q-PCR. Tumor samples with 17q gain as determined by FISH are represented as open bars, whereas those with 17q gain determined by FISH are seen as filled bars. All tumor samples with unbalanced 17q gain by FISH showed relative 17q copy number ratios by Q-PCR of >1.3, with the exception of sample 796.
Primary neuroblastomas with relative 17q copy number values of >1.3 were significantly more likely to be from patients with metastatic disease (Table 3; P < .0001). Concomitant MYCN amplification occurred exclusively in this group of tumors with high relative 17q copy number values (P = .008). These tumors also tended to be from patients older than 1 y of age (P = .07).
DISCUSSION
Neuroblastoma is a clinically heterogeneous disease, and a large number of patients have an outstanding prognosis that can be predicted based on analysis of clinical and tumor biological variables. However, the majority of neuroblastoma cases show hematogenous metastases at diagnosis that is often refractory to treatment (1). Understanding the molecular features that discriminate the unfavorable from the favorable subsets of neuroblastoma has been a focus of intense research in recent years (reviewed in (2)). To date, this research has identified a relatively large panel of somatically acquired genomic alterations that correlate with tumor natural history, including response to treatment. Neuroblastoma has served as a paradigm for the clinical utility of such information, as MYCN oncogene copy number status is used worldwide for risk stratification. Ongoing research is aimed at refining our current risk stratification algorithm by introducing additional tumor-specific prognostic markers that improve our ability to predict tumor behavior.
The current literature supports a strong association between unbalanced gain of chromosome 17q material and an adverse clinical outcome (8, 9, 18, 22, 23, 24). In addition, 17q copy number status may be an independent predictor of outcome even when patient age at diagnosis, disease stage, and MYCN amplification status are considered (9). The major obstacle for incorporating this marker into routine molecular diagnostics is the technical difficulty that is associated with detecting relatively subtle copy number alterations by FISH. This technique is labor intensive, costly, and of low throughput. Therefore, a reliable PCR-based assay that can be used for the high-throughput screening of 17q aberrations in diagnostic neuroblastoma samples would allow for definitive clinical correlative studies aimed at determining the clinical utility of this potential prognostic marker.
Real-time quantitative PCR has been adapted to detect high-level genomic amplification in solid tumor samples, including neuroblastoma (25). However, quantitative assessment of relatively subtle copy number gain in clinical samples has not been reported. The assay described here was designed to provide a relative 17q-17p copy number ratio because unbalanced gain of 17q material has been shown to be prognostically relevant (9, 10, 14, 26, 27). Primer and probe sets were designed based on previous mapping and clinical correlative studies using FISH probes to detect relative abundance of MPO (17q23.1) compared with TP53 (17p13.1) (9, 11). The assay was optimized to allow for use of the comparative Ct method for determining gene copy number, allowing for high throughput by obviating the need for an experiment-specific serial dilution to allow for construction of a standard curve. Data from established cell lines showed the technique to correlate well with interphase FISH data, thus validating the experimental strategy in the artificial setting of uncontaminated cellular material.
For this assay to be of clinical utility, it must be both a sensitive and specific method for detecting unbalanced 17q gain in clinical samples. Neuroblastomas show varying degrees of “contaminating” normal tissues, including vascular and supporting stroma and infiltrating Schwann cells (28). Our data suggest that despite intratumoral cellular heterogeneity, Q-PCR can unequivocally determine 17q allelic status in the majority of clinical samples. In the relatively small sample set studied here, a relative 17q copy number value of 1.3 provided optimal discrimination between tumors with and without unbalanced 17q gain. However, it is also apparent that Q-PCR will provide continuous data across a large sample set, suggesting that this PCR-based technique will not be capable of providing 100% sensitivity and specificity. This should not be surprising, as a significant subset of samples often cannot be reliably classified for 17q status using FISH. Therefore, we plan to score samples with relative 17q copy number values of 1.2–1.4 in future studies as indeterminate by Q-PCR. Each of these samples will then also be examined by dual-color interphase FISH in an attempt to unequivocally assign 17q allelic status for all specimens. Ongoing studies in a much larger number of samples may determine a more precise relative 17q copy number cutoff value and range for samples requiring FISH evaluation.
Q-PCR as described here provides for high throughput screening of relative 17q copy number in primary neuroblastoma specimens. Further studies may refine this assay toward near-perfect sensitivity and specificity, but we expect that there will always be a subset of tumors that should also be studied by FISH if unequivocal determination of 17q allelic status is the goal. Even without further refinement, this assay will allow for highly specific 17q status assignment for the vast majority of neuroblastomas, thus streamlining integration of this molecular marker into clinical correlative studies. Results from prospective evaluation of the clinical significance of 17q aberrations in large numbers of human neuroblastomas may lead to refinements in risk stratification algorithms and thus to more precise allocation to therapeutic protocols.
References
Brodeur GM, Maris JM . Neuroblastoma. In: Pizzo PA, Poplack DG, editors. Principles and practice of pediatric oncology. 4th ed. Philadelphia: Lippincott; 2002. p. 895–938.
Maris JM, Matthay KK . Molecular biology of neuroblastoma. J Clin Oncol 1999; 17: 2264–2279.
Brodeur G, Seeger RC, Schwab M, Varmus HE, Bishop JM . Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984; 224: 1121–1124.
Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med 1985; 313: 1111–1116.
Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B . The International Neuroblastoma Pathology Classification (Shimada) System. Cancer 1999; 86: 364–372.
Look AT, Hayes FA, Shuster JJ, Douglass EC, Castleberry RP, Bowman LC, et al. Clinical relevance of tumor cell ploidy and N-myc gene amplification in childhood neuroblastoma: a pediatric oncology group study. J Clin Oncol 1991; 9: 581–591.
Van Roy N, Laureys G, Cheng NC, Willem P, Opdenakker G, Versteeg R, et al. 1;17 translocations and other chromosome 17 rearrangements in human primary neuroblastoma tumors and cell lines. Genes Chromosomes Cancer 1994; 10: 103–114.
Caron H . Allelic loss of chromosome 1 and additional chromosome 17 material are both unfavourable prognostic markers in neuroblastoma. Med Pediatr Oncol 1995; 24: 215–221.
Bown N, Cotterill S, Lastowska M, O’Neill S, Pearson AD, Plantaz D, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med 1999; 340: 1954–1961.
Lastowska M, Roberts P, Pearson AD, Lewis I, Wolstenholme J, Bown N . Promiscuous translocations of chromosome arm 17q in human neuroblastomas. Genes Chromosomes Cancer 1997; 19: 143–149.
Meddeb M, Danglot G, Chudoba I, Venuat AM, Benard J, Avet-Loiseau H, et al. Additional copies of a 25 Mb chromosomal region originating from 17q23. 1–17qter are present in 90% of high-grade neuroblastomas. Genes Chromosomes Cancer 1996; 17: 156–165.
Savelyeva L, Corvi R, Schwab M . Translocation involving 1p and 17q is a recurrent genetic alteration of human neuroblastoma cells. Am J Hum Genet 1994; 55: 334–340.
McConville CM, Dyer S, Rees SA, Luttikhuis ME, McMullan DJ, Vickers SJ, et al. Molecular cytogenetic characterization of two non-MYCN amplified neuroblastoma cell lines with complex t(11;17). Cancer Genet Cytogenet 2001; 130: 133–140.
Stark B, Jeison M, Bar-Am I, Glaser-Gabay L, Mardoukh J, Luria D, et al. Distinct cytogenetic pathways of advanced-stage neuroblastoma tumors, detected by spectral karyotyping. Genes Chromosomes Cancer 2002; 34: 313–324.
Brodeur GM, Pritchard J, Berthold F, Carlsen NLT, Castel V, Castleberry RP, et al. Revisions in the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 1993; 11: 1466–1477.
Mathew P, Valentine MB, Bowman LC, Rowe ST, Nash MB, Valentine VA, et al. Detection of MYCN gene amplification in neuroblastoma by fluorescence in situ hybridization: a pediatric oncology group study. Neoplasia 2001; 3: 105–109.
Plantaz D, Mohapatra G, Matthay KK, Pellarin M, Seeger RC, Feuerstein BG . Gain of chromosome 17 is the most frequent abnormality detected in neuroblastoma by comparative genomic hybridization. Am J Pathol 1997; 150: 81–89.
Brinkschmidt C, Christiansen H, Terpe HJ, Simon R, Lampert F, Boecker W, et al. Distal chromosome 17 gains in neuroblastomas detected by comparative genomic hybridization (CGH) are associated with a poor clinical outcome. Med Pediatr Oncol 2001; 36: 11–13.
Lastowska M, Cotterill S, Bown N, Cullinane C, Variend S, Lunec J, et al. Breakpoint position on 17q identifies the most aggressive neuroblastoma tumors. Genes Chromosomes Cancer 2002; 34: 428–436.
Trakhtenbrot L, Cohen N, Betts DR, Niggli FK, Amariglio N, Brok-Simoni F, et al. Interphase fluorescence in situ hybridization detection of chromosome 17 and 17q region gains in neuroblastoma: are they secondary events? Cancer Genet Cytogenet 2002; 137: 95–101.
Applied Biosystems. ABI PRISM 7700 Sequence Detection System User Guide. In: User Bulletin #2: Applied Biosystems.
Abel F, Ejeskar K, Kogner P, Martinsson T . Gain of chromosome arm 17q is associated with unfavourable prognosis in neuroblastoma, but does not involve mutations in the somatostatin receptor 2(SSTR2) gene at 17q24. Br J Cancer 1999; 81: 1402–1409.
Lastowska M, Cotterill S, Pearson AD, Roberts P, McGuckin A, Lewis I, et al. Gain of chromosome arm 17q predicts unfavourable outcome in neuroblastoma patients.U.K. Children’s Cancer Study Group and the U.K. Cancer Cytogenetics Group. Eur J Cancer 1997; 33: 1627–1633.
Lastowska M, Cullinane C, Variend S, Cotterill S, Bown N, O’Neill S, et al. Comprehensive genetic and histopathologic study reveals three types of neuroblastoma tumors. J Clin Oncol 2001; 19: 3080–3090.
De Preter K, Speleman F, Combaret V, Lunec J, Laureys G, Eussen BH, et al,. Quantification of MYCN, DDX1, and NAG gene copy number in neuroblastoma using a real-time quantitative PCR assay. Mod Pathol 2002; 15: 159–166.
Cunsolo CL, Bicocchi MP, Petti AR, Tonini GP . Numerical and structural aberrations in advanced neuroblastoma tumours by CGH analysis; survival correlates with chromosome 17 status. Br J Cancer 2000; 83: 1295–1300.
Lastowska M, Van Roy N, Bown N, Speleman F, Lunec J, Strachan T, et al. Molecular cytogenetic delineation of 17q translocation breakpoints in neuroblastoma cell lines. Genes Chromosomes Cancer 1998; 23: 116–122.
Ambros IM, Zellner A, Roald B, Amann G, Ladenstein R, Printz D, et al. Role of ploidy, chromosome 1p, and Schwann cells in the maturation of neuroblastoma. N Engl J Med 1996; 334: 1505–1511.
Acknowledgements
This work was supported in part by and NIH Grants R01-CA87847 (J.M.M.) and U01-CA30969 (Children’s Oncology Group), and a Hope Street Kids Cancer Research Foundation Grant (Q.W. and J.M.M). The authors would like to thank Ms. Luanne Wainwright for technical assistance and Dr. Hiro Shimada for providing the central review of neuroblastoma histopathology.
Authors Morowitz and Shusterman contributed equally to this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Morowitz, M., Shusterman, S., Mosse, Y. et al. Detection of Single-Copy Chromosome 17q Gain in Human Neuroblastomas Using Real-Time Quantitative Polymerase Chain Reaction. Mod Pathol 16, 1248–1256 (2003). https://doi.org/10.1097/01.MP.0000097364.64566.81
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000097364.64566.81
Keywords
This article is cited by
-
Mechanisms of invasion and metastasis in human neuroblastoma
Cancer and Metastasis Reviews (2007)
-
cDNA array-CGH profiling identifies genomic alterations specific to stage and MYCN-amplification in neuroblastoma
BMC Genomics (2004)
