
Abstract
Experimental and observational evidence suggests that chronic hypoxic stimulation can induce parasympathetic paraganglioma. This is emphasized by the identification of germline mutations in genes of the mitochondrial succinate dehydrogenase enzyme complex II in hereditary paraganglioma. Because of inactivating mutations in the succinate dehydrogenase subunit B (SDHB), C (SDHC), or D (SDHD) gene, the paraganglia undergo a chronic hypoxic stimulus leading to proliferation of the paraganglionic cells. Hypoxia is a known inducer of p53 up-regulation, which triggers cell cycle arrest and apoptosis. Inactivation of the p53 pathway, by gene mutation or by MDM2 overexpression, would enable cells to escape from cell cycle arrest and apoptosis and could contribute to tumorigenesis. To determine whether p53 inactivation plays a role in paraganglioma tumorigenesis, we investigated a series of 43 paragangliomas from 41 patients (of whom 24 patients harbored a germline SDHD mutation) for mutations in p53 exons 5–8 by PCR-SSCP. In addition, these tumors were investigated for p53 and MDM2 protein expression by immunohistochemistry, and the results were compared with clinical data and the presence of SDHD mutations. No aberrations in p53 exons 5–8 were found. The immunohistochemical experiments showed nuclear p53 expression in 15 tumors. Three tumors were positive for MDM2 that were also positive for p53. There was no correlation between p53 and MDM2 expression and clinical data or SDHD status. Given the fact that hypoxia induces p53 expression and regarding the absence of p53 mutations, these results suggest that p53 inactivation does not play a major role in the tumorigenesis of hereditary and sporadic paragangliomas.
Similar content being viewed by others

INTRODUCTION
Parasympathetic paragangliomas (OMIM #168000) originate from neural crest–derived chief cells in the paraganglia. The tumors occur mostly in the head and neck region, with the carotid body being the most frequent location of paragangliomas, followed by the jugulotympanic paraganglia. The tumors are slowly growing, highly vascularized, and mostly benign, but metastatic spread is found in ∼10% of patients (reviewed in 1).
A positive family history is present in 10 to 50% of the patients (2, 3, 4), but genetic predisposition may also be present in 8 to 32% of isolated patients (5, 6). Genetic predisposition to parasympathetic paraganglioma was recently revealed by the identification of germline mutations in subunit D of the mitochondrial succinate dehydrogenase enzyme complex II (SDHD) in familial paraganglioma patients (7). Since then, mutations in other subunits, B (SDHB) and C (SDHC) of complex II, have also been found to predispose to paraganglioma development (8, 9). Co-occurrence of parasympathetic paragangliomas and their sympathoadrenal counterpart pheochromocytomas and association with Carney's syndrome and neurofibromatosis type 1 has been described (10, 11, 12, 13).
Apart from mutations in succinate dehydrogenase enzyme complex II, little is known about the pathogenetic mechanisms underlying paraganglioma development. By comparative genomic hybridization, we previously detected that loss of chromosome 11 is the only recurrent chromosomal aberration in parasympathetic paragangliomas, particularly in familial paragangliomas (14). Overall DNA copy number changes are infrequent, which is in concordance with the benign and slow-growing nature of these tumors. Flow cytometric analyses revealed DNA aneuploidy in 21–50% of the tumors, which was not predictive of malignant behavior or decreased survival (15, 16, 17). A few immunohistochemical studies have suggested a paracrine/autocrine role for IGF-II, c-myc, bcl-2, and c-jun in paraganglioma pathogenesis (18, 19, 20, 21).
The mitochondrial succinate dehydrogenase enzyme complex II is involved in the citric acid cycle and the aerobic respiratory chain (22). A complete loss of complex II enzymatic activity, due to inactivating mutations in the SDHB, SDHC, or SDHD gene and loss of heterozygosity of the corresponding wild type allele, leads to a high expression of hypoxic-angiogenic responsive genes like vascular endothelial growth factor (VEGF) and endothelial periodic acid–Schiff domain protein 1 (EPAS1/HIF2α) (23, 24). The fact that cellular hypoxia stimulates paraganglioma development is further suggested by a markedly increased incidence of carotid body paragangliomas in people living permanently under hypoxic conditions (at high altitude or because of chronic obstructive pulmonary disease) (25, 26, 27). Cellular stress such as DNA damage or hypoxia induces p53 (28), after which MDM2 is up-regulated to serve as negative feedback for p53. Induction of the tumor suppressor gene p53 results in cell cycle arrest at the G0-G1 boundary, but when p53 is mutated, control of cell proliferation is lost. Cells with mutated p53 have a growth advantage compared with the surrounding cells, and this can contribute to tumor formation. Obviously, paraganglioma cells escape from hypoxia-induced cellular senescence. One of the mechanisms for circumventing the hypoxia-induced cellular senescence is the inactivation of p53. In numerous tumor types, p53 inactivation is caused by mutation in the p53 gene itself or by MDM2 overexpression (29, 30). The MDM2 protein targets p53 for proteasomal degradation and is as such involved in the perturbation of p53 function (31, 32). There is strong evidence that p53 mutation and MDM2 overexpression are mutually exclusive in most tumors and represent two alternative mechanisms to inactivate suppression of cell growth.
In paragangliomas, investigations on p53 alterations are scarce, and molecular analysis especially is lacking (33, 34, 35). These data prompted us to determine the expression of p53 and MDM2 in a series of hereditary and sporadic paragangliomas. In addition, p53 exons 5–8 were investigated for mutations by PCR-SSCP.
METHODS
Patients and Tumor Samples
From our archival files, we randomly selected 43 parasympathetic paragangliomas from 41 patients who were diagnosed between 1987 and 2000 at the Erasmus Medical Center, Rotterdam, The Netherlands (see Table 1). Of these patients, 24 were female and 17 were male. The mean age was 42 years (range, 20–74 y), and 17 patients (41%) had a positive family history. SDHD mutation analysis had been performed previously in all patients, and germline mutations were found in 24 (59%) patients: 16 patients had the Dutch founder mutation D92Y, 6 patients harbored the L95P mutation, and in 2 patients, the L139P mutation was found (6). Table 1 summarizes all relevant clinical characteristics of the 41 paraganglioma patients evaluated for p53/MDM2 alterations in this study.
DNA Isolation
DNA was isolated from frozen (n = 7) or (n = 36) paraffin-embedded tissues. Tissue regions consisting of ≥80% neoplastic cells were selected from H&E–stained sections. These regions were manually dissected from (deparaffinized) unstained consecutive sections. White blood cell pellets from healthy volunteer blood donors and cell pellets from cultured tumor cells were used as controls. Dissected tissue fragments and the cell pellets were digested overnight at 56° C in 200 μL of digestion buffer containing 10 μL of Proteinase K (20 μg/μL), 50 mmol/L Tris-HCL (pH 8.0), 100 mmol/L EDTA, and 0.5% sodium dodecyl sulfate. DNA was extracted by phenol-chloroform and precipitated with ethanol. Pellets were dissolved in 10 mm Tris-HCL (pH 7.8).
PCR-SSCP
Exons 5 to 8 of the p53 gene, including the exon-intron boundaries, were investigated by PCR-SSCP. As controls, DNA samples from normal individuals were used. In addition, DNA from the prostate carcinoma cell lines PC-3 and Du-145, and from the colorectal carcinoma cell lines Colo-320 and HT-29, with known p53 mutations in exons 5, 6, 7, and 8, respectively, served as positive controls. The DNA isolated from routine formalin-fixed and paraffin-embedded tissues is highly degraded; therefore, we used small-amplicon (<200 bp) PCR to investigate exons 5–8 of the p53 gene. All four exons were amplified in two fragments each, as recently described (36). PCR was performed in 15-μL reaction volume consisting of (per 50 μL): 1 U Taq DNA polymerase (Promega, Madison, WI), 1.5 mm MgCl2, 200 ng of each primer, 0.2 mm dGTP, dTTP, dCTP, 0.02 mm dATP, 2.5 μCi α-32P-dATP, and approximately 100 ng of DNA. Temperatures for amplification were 95° C for 30 seconds, 55° C for 45 seconds, and 72° C for 45 seconds. These steps were repeated for 35 cycles, followed by a final extension at 72° C for 10 minutes. The PCR product was diluted with an equal amount of loading buffer (95% formamide; 10 mm EDTA, pH 8.0; 0.025% bromophenol blue; and 0.025% xylene cyanol) and denaturated at 95° C for 5 minutes. The solution was chilled on ice, and 4 μL was loaded on a 8% polyacrylamide gel (acrylamide to bisacrylamide, 49:1) containing 10% glycerol. Electrophoresis was performed at 8W for 16 hours at room temperature. Gels were vacuum dried at 80° C and exposed to X-ray films.
Immunohistochemistry
Five-micrometer sections of paraffin-embedded tumors were mounted onto amino-alkyl-silane–coated slides and deparaffinized. Subsequently, slides were washed twice in 100% alcohol, incubated for 20 minutes in 3% H2O2 in methanol, and rinsed with tap water. A microwave antigen retrieval method (15 min in citrate buffer, pH 6, at 600 W) was used, followed by incubation for 15 minutes in 10% normal goat serum (DAKO, Glostrup, Denmark). Do7 anti-p53 monoclonal antibody (DAKO) was used at a dilution of 1:50 for 30 minutes at room temperature, and the MDM2 monoclonal antibody 1B10 (Novocastra Laboratories, Newcastle upon Tyne, United Kingdom) was used at a dilution of 1:25 for 30 minutes at room temperature, both followed by biotinylated goat-anti-multilink and streptavidin-biotin peroxidase complex (both undiluted; Lab Vision Corporation, Fremont, CA). Visualization was achieved by diaminobenzidine tetrahydrochloride (Fluka, Neu-Ulm, Germany) with 3% H2O2 for 7 minutes.
In the negative control reactions, the primary antibodies were omitted from the dilution buffer (phosphate-buffered saline with 5% bovine serum albumin). A p53-positive esophageal adenocarcinoma and an MDM2-positive breast carcinoma were used as positive controls.
Staining of p53 and MDM2 was assessed according to the method described by Sinicrope et al. (37). This method is based on the percentage of positive tumor cells and the staining intensity. A score of 0 to 4 was assigned according to the percentage of positively stained tumor cells: 0 = positive staining in <5%; 1 = >5–25%; 2 = >25–50%; 3 = >50–75%; and 4 = >75%. These results are multiplied by the staining intensity score of the tumor cells: 1 = negative–weak; 2 = moderate; and 3 = strong staining. A multiplied score of ≥6 is regarded as positive staining, and a score of <6, as negative.
Statistics
Correlations between p53 and MDM2 alterations and SDHD mutation status or clinical features were tested by use of the χ2 test or of an unpaired t test. P values of < .05 were considered statistically significant.
RESULTS
PCR-SSCP Analysis
PCR products of p53 exon 5–8 could be obtained from all 43 tumor/normal DNA samples. By SSCP analysis, no aberrations were found in the 43 tumor samples, whereas the four different p53 mutations in the tumor cell lines were clearly identified with the applied SSCP conditions. Figure 1 shows an example of a PCR-SSCP normal pattern of paraganglioma samples and a band shift of a positive control (PC-3). This cell line contained a C deletion in codon 138 of the p53 gene.

Examples of SSCP analysis of p53 exon 5 and exon 7 in parasympathetic paragangliomas. The autoradiographs of the PCR-SSCP gel show the migration patterns of tumor (T) and normal (N) DNA and the mobility shifts (red arrowheads) produced by aberrant control samples (C) of the positive controls PC3 (exon 5) and Colo-320 (exon 7).
p53/MDM2 Protein Expression and Association with SDHD Mutations
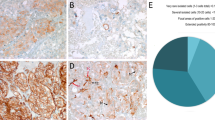
Of 43 paragangliomas, p53 immunoreactivity was detected in 15 tumors (35%) of 13 patients. Three tumors (7%) from different patients showed concurrent MDM2 expression, leaving the majority (n = 28, 65%) of the tumors negative for both p53 and MDM2. Immunoreactivity of p53 and MDM2 was observed both in the nucleus and the cytoplasm. Also, p53 positivity was observed in tumor and stromal cells in all these cases. Figure 2 shows examples of positive and negative staining of p53 and MDM2.

Immunohistochemical staining of p53 and MDM2 in parasympathetic paragangliomas using the anti-p53 monoclonal antibody Do7 and MDM2 monoclonal antibody 1B10, respectively. A, positive p53 staining (left) of tumor and stromal cells in PGL20, a mediastinal paraganglioma of a patient with a negative family history and no germline SDHD mutation. MDM2 expression is absent in PGL20 (right). B, PGL3, a vagal paraganglioma of a patient with multiple paragangliomas, a positive family history, and a germline D92Y SDHD mutation. Tumor and stromal cells stain positive for p53, whereas MDM2 staining is mainly present in the tumor cells. Note the nuclear and cytoplasmic staining of p53 in both paragangliomas and the cytoplasmic presence of MDM2 in the tumor cells of PGL3.
From a patient with bilateral carotid body tumors, one tumor was p53 positive, whereas the other tumor was p53 negative. A vagal and a carotid body tumor of another patient both showed the same expression pattern (p53+/MDM2−). Of the 13 patients with a p53-positive paraganglioma, 9 had a single paraganglioma, 4 of which recurred after resection. The other 4 paragangliomas with detectable p53 were from patients with bilateral or multiple tumors. There was no correlation between p53/MDM2 status and tumor focality or tumor location.
Because hypoxia is known to be present in SDHD-mutated paraganglionic cells and hypoxia is known to stimulate p53 transcription, leading to cell cycle arrest and apoptosis, abrogation of the p53 pathway could especially be expected in SDHD-mutated paragangliomas. However, p53 positivity was present in 6 of 25 (24%) tumors with an SDHD mutation, and 9 of 18 (50%) tumors without an SDHD mutation were positive for p53. Similarly, MDM2-positive staining was found in one patient with an SDHD mutation.
By calculating the significance of the correlation of p53 expression with sex, family history, tumor focality (follow-up), site of the tumor, and SDHD germline status, none of these parameters was shown to be significantly associated with absence of p53 immunoreactivity. Results of p53 and MDM2 immunotyping and correlations with tumor and patient characteristics are shown in Table 2.
DISCUSSION
Experimental and observational evidence indicates that chronic hypoxic stimulation is involved in the tumorigenesis of paraganglioma. Hypoxia is a well-known inducer of p53, which in turn results in cell cycle arrest or apoptosis, a mechanism that is abrogated in most, if not all, cancers. The present study was undertaken to investigate the possible involvement of p53 in the development of parasympathetic paragangliomas with and without SDHD mutations, using immunohistochemical assessment of p53 and MDM2 expression and mutation analysis of p53 exon 5–8.
Fifteen of the 43 investigated paragangliomas (35%) showed nuclear and cytoplasmic p53 immunoreactivity. MDM2 staining was observed in 3 tumors (7%) that were simultaneously positive for p53. We found a p53-MDM2 concordance of 75%, similar to that described in breast and colorectal carcinoma (38, 39). p53 immunoreactivity was more frequent in paragangliomas without SDHD mutations (50%) than in paragangliomas with SDHD mutations (24%), although this was not statistically significant (P = .08).
Under normal conditions, the p53 concentration in cells is low and cannot be detected by immunohistochemistry. Through cellular stress, the concentration of p53 can rise and hence be detected by immunohistochemistry (40, 41). In addition, mutant p53 often has a longer half-life than wild type p53 and can be detected immunohistochemically (42, 43). However, there is no direct correlation between p53 mutation and immunohistochemical p53 overexpression (32, 44). The immunohistochemical detection of p53 expression in 15 paragangliomas indicates increased wild type p53 expression or the presence of mutant p53. However, no aberrations in exons 5–8 of the p53 gene were found by PCR-SSCP. It is known from the literature that >95% of p53 mutations are found in exons 5–8 (29), but we cannot exclude the presence of mutations outside this region. In addition, the mutation detection efficiency of PCR-SSCP is not 100%, and mutations could remain undetected, although all four different control p53 mutations were identified by the procedure used. Despite this, we consider our molecular results to be strong indication that p53 mutations do not contribute to paraganglioma tumorigenesis. Moreover, the observation of p53 immunoreactivity in tumor and stromal cells suggests hypoxia rather than gene mutation as the cause of p53 expression. Inactivation of p53 in tumors is often the result of the combination of a mutant p53 allele and 17p allele loss. In several molecular studies, no 17p loss in paragangliomas has been found (14, 45). This is in accordance with the observed absence of p53 mutations in these tumors. A recent investigation has shown that the increase in p53 during hypoxia is not accompanied by a parallel rise in MDM2 (40). If p53 is active in the p53-expressing paragangliomas, this implies that the tumorigenic mechanism in these tumors overrules the tumor suppressor capacity of wild type p53. In accordance with this concept, paragangliomas are very slowly growing tumors.
MDM2 overexpression in tumors with wild type p53 accumulation has also been described in bladder, testicular, esophageal, and laryngeal carcinoma and in acute lymphoblastic leukemia (46, 47, 48, 49, 50). As suggested in the literature, the concomitant expression of MDM2 and p53 proteins indicates inactive p53, implying that p53 is inactive in the three paragangliomas with MDM2 expression in this study. In the remaining 12 p53-positive paragangliomas, p53 could be active, although inactivation of p53 by other proteins like viral oncogenes or cellular proteins cannot be excluded (51).
In 28 (65%) of the investigated paragangliomas, besides the absence of p53 mutations, no p53 expression was detected. This could point to a p53-independent tumorigenic pathway. Nineteen of these 28 tumors have an SDHD gene mutation resulting in cellular hypoxia. Obviously, hypoxia in these tumors does not lead to p53 up-regulation. However, there are ways to perturb the p53 pathway during tumor development in addition to the commonly seen p53 gene mutations or MDM2 overexpression. These include loss of the ability to stabilize p53 through mechanisms such as loss of ARF or inactivation of kinases, inappropriate localization of p53, and inactivation of downstream mediators of p53 such as Apaf-1 or Bax (52, 53). Many cancers with wild type p53 show loss of the p14ARF protein, resulting in destabilization of p53 (54). This loss is often the result of p14ARF locus deletion, but in paragangliomas, loss of chromosomal region 9p has not been observed (14, 45). Also, in a case report of two brothers with paraganglioma, no allele loss nor mutations in p53 and the 9p gene p16INK4A were found (55). More than 8 years after radiotherapy, a recurrence appeared to have a p53 as well as a p16INK4A mutation, and those investigators suggest that these mutations may have resulted from the therapy.
In summary, our data indicate that p53 is expressed in ≥35% of paragangliomas, independently of SDHD gene status and not caused by p53 gene mutations. Abrogation of the p53 tumor surveillance mechanism by MDM2 overexpression is detected in a small subset (7%) of these tumors, which also is not associated with SDHD gene mutations. Further experiments are needed to clarify the mechanisms by which paragangliomas escape from apoptotic signals.
References
Lee JH, Barich F, Karnell LH, et al. National Cancer Data Base report on malignant paragangliomas of the head and neck. Cancer 2002; 94: 730–7.
Grufferman S, Gillman MW, Pasternak LR, Peterson CL, Young WG . Familial carotid body tumors: case report and epidemiologic review. Cancer 1980; 46: 2116–22.
Hodge KM, Byers RM, Peters LJ . Paragangliomas of the head and neck. Arch Otolaryngol Head Neck Surg 1988; 114: 872–7.
van Baars F, Cremers C, van den Broek P, Geerts S, Veldman J . Genetic aspects of nonchromaffin paraganglioma. Hum Genet 1982; 60: 305–9.
Baysal BE, Willett-Brozick JE, Lawrence EC, et al. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet 2002; 39: 178–83.
Dannenberg H, Dinjens WN, Abbou M, et al. Frequent germ-line succinate dehydrogenase subunit D gene mutations in patients with apparently sporadic parasympathetic paraganglioma. Clin Cancer Res 2002; 8: 2061–6.
Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000; 287: 848–51.
Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 2001; 69: 49–54.
Niemann S, Muller U . Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet 2000; 26: 268–70.
Jensen JC, Choyke PL, Rosenfeld M, et al. A report of familial carotid body tumors and multiple extra-adrenal pheochromocytomas. J Urol 1991; 145: 1040–2.
Pritchett JW . Familial concurrence of carotid body tumor and pheochromocytoma. Cancer 1982; 49: 2578–9.
DeAngelis LM, Kelleher MB, Post KD, Fetell MR . Multiple paragangliomas in neurofibromatosis: a new neuroendocrine neoplasia. Neurology 1987; 37: 129–33.
Carney JA . Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc 1999; 74: 543–52.
Dannenberg H, de Krijger RR, Zhao J, et al. Differential loss of chromosome 11q in familial and sporadic parasympathetic paragangliomas detected by comparative genomic hybridization. Am J Pathol 2001; 158: 1937–42.
Barnes L, Taylor SR . Vagal paragangliomas: a clinical, pathological, and DNA assessment. Clin Otolaryngol 1991; 16: 376–82.
Sauter ER, Hollier LH, Bolton JS, Ochsner JL, Sardi A . Prognostic value of DNA flow cytometry in paragangliomas of the carotid body. J Surg Oncol 1991; 46: 151–3.
van der Mey AG, Cornelisse CJ, Hermans J, Terpstra JL, Schmidt PH, Fleuren GJ . DNA flow cytometry of hereditary and sporadic paragangliomas (glomus tumours). Br J Cancer 1991; 63: 298–302.
Wang DG, Barros D'Sa AA, Johnston CF, Buchanan KD . Oncogene expression in carotid body tumors. Cancer 1996; 77: 2581–7.
Jyung RW, LeClair EE, Bernat RA, et al. Expression of angiogenic growth factors in paragangliomas. Laryngoscope 2000; 110: 161–7.
Li SL, Goko H, Xu ZD, et al. Expression of insulin-like growth factor (IGF)-II in human prostate, breast, bladder, and paraganglioma tumors. Cell Tissue Res 1998; 291: 469–79.
Wang DG, Johnston CF, Barros D'Sa AA, Buchanan KD . Expression of apoptosis-suppressing gene bcl-2 in human carotid body tumours. J Pathol 1997; 183: 218–21.
Hirawake H, Taniwaki M, Tamura A, Amino H, Tomitsuka E, Kita K . Characterization of the human SDHD gene encoding the small subunit of cytochrome b (cybS) in mitochondrial succinate-ubiquinone oxidoreductase. Biochim Biophys Acta 1999; 1412: 295–300.
Gimenez-Roqueplo AP, Favier J, Rustin P, et al. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet 2001; 69: 1186–97.
Gimenez-Roqueplo AP, Favier J, Rustin P, et al. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J Clin Endocrinol Metab 2002; 87: 4771–4.
Saldana MJ, Salem LE, Travezan R . High altitude hypoxia and chemodectomas. Hum Pathol 1973; 4: 251–63.
Pacheco-Ojeda L, Durango E, Rodriquez C, Vivar N . Carotid body tumors at high altitudes: Quito, Ecuador, 1987. World J Surg 1988; 12: 856–60.
Chedid A, Jao W . Hereditary tumors of the carotid bodies and chronic obstructive pulmonary disease. Cancer 1974; 33: 1635–41.
Greenblatt MS, Bennett WP, Hollstein M, Harris CC . Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res 1994; 54: 4855–78.
Hollstein M, Sidransky D, Vogelstein B, Harris CC . p53 mutations in human cancers. Science 1991; 253: 49–53.
Deb SP . Function and dysfunction of the human oncoprotein MDM2. Front Biosci 2002; 7: d235–43.
Kubbutat MH, Jones SN, Vousden KH . Regulation of p53 stability by Mdm2. Nature 1997; 387: 299–303.
Haupt Y, Maya R, Kazaz A, Oren M . Mdm2 promotes the rapid degradation of p53. Nature 1997; 387: 296–9.
Lam KY, Lo CY, Wat NM, Luk JM, Lam KS . The clinicopathological features and importance of p53, Rb, and mdm2 expression in phaeochromocytomas and paragangliomas. J Clin Pathol 2001; 54: 443–8.
Welkoborsky HJ, Gosepath J, Jacob R, Mann WJ, Amedee RG . Biologic characteristics of paragangliomas of the nasal cavity and paranasal sinuses. Am J Rhinol 2000; 14: 419–26.
Cheng L, Leibovich BC, Cheville JC, et al. Paraganglioma of the urinary bladder: can biologic potential be predicted? Cancer 2000; 88: 844–52.
van der Sijp JR, van Meerbeeck JP, Maat AP, et al. Determination of the molecular relationship between multiple tumors within one patient is of clinical importance. J Clin Oncol 2002; 20: 1105–14.
Sinicrope FA, Ruan SB, Cleary KR, Stephens LC, Lee JJ, Levin B . bcl-2 and p53 oncoprotein expression during colorectal tumorigenesis. Cancer Res 1995; 55: 237–41.
Elkablawy MA, Maxwell P, Williamson K, Anderson N, Hamilton PW . Apoptosis and cell-cycle regulatory proteins in colorectal carcinoma: relationship to tumour stage and patient survival. J Pathol 2001; 194: 436–43.
McCann AH, Kirley A, Carney DN, et al. Amplification of the MDM2 gene in human breast cancer and its association with MDM2 and p53 protein status. Br J Cancer 1995; 71: 981–5.
Koumenis C, Alarcon R, Hammond E, et al. Regulation of p53 by hypoxia: dissociation of transcriptional repression and apoptosis from p53-dependent transactivation. Mol Cell Biol 2001; 21: 1297–310.
Ashcroft M, Taya Y, Vousden KH . Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol 2000; 20: 3224–33.
Baas IO, Mulder JW, Offerhaus GJ, Vogelstein B, Hamilton SR . An evaluation of six antibodies for immunohistochemistry of mutant p53 gene product in archival colorectal neoplasms. J Pathol 1994; 172: 5–12.
Gannon JV, Greaves R, Iggo R, Lane DP . Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. EMBO J 1990; 9: 1595–602.
Battifora H . p53 immunohistochemistry: a word of caution. Hum Pathol 1994; 25: 435–37.
Devilee P, van Schothorst EM, Bardoel AF, et al. Allelotype of head and neck paragangliomas: allelic imbalance is confined to the long arm of chromosome 11, the site of the predisposing locus PGL. Genes Chromosomes Cancer 1994; 11: 71–8.
Lianes P, Orlow I, Zhang ZF, et al. Altered patterns of MDM2 and TP53 expression in human bladder cancer. J Natl Cancer Inst 1994; 86: 1325–30.
Zhou M, Yeager AM, Smith SD, Findley HW . Overexpression of the MDM2 gene by childhood acute lymphoblastic leukemia cells expressing the wild-type p53 gene. Blood 1995; 85: 1608–14.
Riou G, Barrois M, Prost S, Terrier MJ, Theodore C, Levine AJ . The p53 and mdm-2 genes in human testicular germ-cell tumors. Mol Carcinog 1995; 12: 124–31.
Pruneri G, Pignataro L, Carboni N, et al. MDM-2 oncoprotein overexpression in laryngeal squamous cell carcinoma: association with wild-type p53 accumulation. Mod Pathol 1997; 10: 785–92.
Soslow RA, Altorki NK, Yang G, Xie D, Yang CS . mdm-2 expression correlates with wild-type p53 status in esophageal adenocarcinoma. Mod Pathol 1999; 12: 580–6.
Pise-Masison CA, Radonovich M, Sakaguchi K, Appella E, Brady JN . Phosphorylation of p53: a novel pathway for p53 inactivation in human T-cell lymphotropic virus type 1-transformed cells. J Virol 1998; 72: 6348–55.
Soengas MS, Capodieci P, Polsky D, et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 2001; 409: 207–11.
Rampino N, Yamamoto H, Ionov Y, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997; 275: 967–9.
Sherr CJ, Weber JD . The ARF/p53 pathway. Curr Opin Genet Dev 2000; 10: 94–9.
Guran S, Tali ET . p53 and p16INK4A mutations during the progression of glomus tumor. Pathol Oncol Res 1999; 5: 41–5.
Acknowledgements
Supported by a grant of the Vanderes Foundation.
The authors thank Frank van der Panne for assistance in the generation of the figures.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
van Nederveen, F., Dannenberg, H., Sleddens, H. et al. p53 Alterations and Their Relationship to SDHD Mutations in Parasympathetic Paragangliomas. Mod Pathol 16, 849–856 (2003). https://doi.org/10.1097/01.MP.0000084111.03922.4D
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000084111.03922.4D
