
Abstract
Cyclins play an important role in regulating the passage of dividing cells through critical checkpoints in the cell cycle. Because alterations of several cyclins, especially cyclin D1, have been implicated in the development of many human neoplasms, we examined 32 cases of giant cell tumor of long bones for cyclin D1 gene amplification and protein overexpression using differential polymerase chain reaction and immunohistochemistry, respectively. In addition, the expression of cyclin D3, cyclin B1, and the proliferation-associated antigen Ki-67 (MIB-1) was assessed immunohistochemically. Low-level cyclin D1 gene amplification was detected in 61% of giant cell tumor cases. All tumors showed cyclin D1, cyclin D3, cyclin B1, and Ki-67 (MIB-1) staining; however, the distribution was very characteristic. Cyclin D1 protein expression was seen predominantly in the nuclei of the giant cells, with occasional mononuclear cells staining. There was no correlation between cyclin D1 gene amplification and protein overexpression. Cyclin D3 staining showed a similar distribution, with 88% of cases showing protein overexpression. Cyclin D1 and/or D3 staining in the giant cells was never associated with staining for either cyclin B1 or Ki-67 (MIB-1), as the expression of the latter two proteins was restricted to the mononuclear cells. Cyclin B1 overexpression was seen in 44% of cases. Ki-67 (MIB-1) staining was present in all cases, and between 10 to 50% of the mononuclear cells were positive. These results suggest that alterations in cyclin D1 and/or D3 might play a role in the pathogenesis of giant cell tumor of bone.
Similar content being viewed by others
INTRODUCTION
Alterations of several cell cycle regulatory proteins, especially those involved in the G1-to-S transition, such as D-type cyclins, are detected in many human tumors. Genes encoding D-type cyclins (D1, D2, and D3) are induced by mitogenic stimuli (1) and, together with their catalytic partners CDK4 and CDK6, phosphorylate the retinoblastoma protein (pRb) (2) and allow cells to cross the G1/S restriction point. The cells then become committed to DNA replication and completion of the cell cycle (3). Because D-type cyclins provide the link between mitogenic signals and activation of the cell cycle (1, 4) and because of their regulatory function in the G1-to-S transition (5), constitutive activation of the D-cyclin pathway can reduce or overcome certain mitogen requirements for cell proliferation (6) and thereby contribute to oncogenic transformation (7, 8, 9). Although studies suggest that overexpression of cyclin D1 appears to be insufficient on its own to transform primary cells, it may cooperate with other oncogenes to induce transformation (10, 11, 12, 13). Amplification of the cyclin D1 gene and/or overexpression of the protein are observed in a number of human malignancies, such as certain types of epithelial cancers, lymphomas, and some central nervous system tumors (14). Benign and premalignant lesions of the breast (15) and epithelial dysplasias of the oral and laryngeal mucosa (16, 17) also show cyclin D1 alterations, suggesting that these changes occur before malignant transformation.
Whereas D-type cyclins are involved in the G1-to-S transition, B-type cyclins are implicated in the control of the G2-to-M transition and mitosis. Activation of the cyclin B/CDK1 complex triggers entry into mitosis in all eukaryotic cells. Cyclin B2/CDK1 associates with the Golgi apparatus (18) and plays a role in Golgi remodeling during mitosis (19). Cyclin B1 localization changes dramatically during the cell cycle. It accumulates in the cytoplasm during S and G2 phases, then moves into the nucleus at the G2/M transition to phosphorylate substrates critical for mitosis (20). An increasing body of data suggests that altered expression of cyclin B1 occurs in human neoplasia, and overexpression of cyclin B1 has been demonstrated in lung, colorectal, prostate, breast, esophagus, and head and neck cancers (21, 22, 23, 24, 25, 26, 27).
Giant cell tumor of bone is an uncommon primary bone tumor that occurs in young adults and predominantly affects the ends of long tubular bones, most notably the distal femur and proximal tibia, where 50% of cases occur. Giant cell tumor can be locally aggressive, with a tendency for recurrence. Lung metastases are described and occur in 1 to 2% of patients with giant cell tumor (28). Given the proliferative activity detected in giant cell tumor (29, 30, 31, 32, 33), it is possible that there is deregulation of the cell cycle at either the G1/S or the G2/M transition. To investigate for these alterations, 32 cases of giant cell tumor of bone were examined for cyclin D1 gene amplification and/or protein overexpression using differential polymerase chain reaction and immunohistochemistry. In addition, the expression of cyclin D3, cyclin B1, and the proliferation-associated antigen Ki-67 was assessed immunohistochemically.
MATERIALS AND METHODS
Tissues and Cell Lines
Forty-six formalin-fixed, paraffin-embedded tissue blocks coded as giant cell tumor of bone from 41 patients were retrieved from the files of the Department of Pathology and Laboratory Medicine, Mount Sinai Hospital, Toronto, from 1991 to 2001. Hematoxylin and eosin-stained slides were reviewed by two of the authors (AK, RK) to confirm the diagnosis and evaluate the quality of the tissue. For inclusion in the study, the tumors had to be primary (nonrecurrent), and the tissue had to be non-decalcified.
The ZR-75-1 cell line, which has a twofold to fivefold cyclin D1 gene amplification (34, 35), and the MDA-MB-231 cell line, which has no cyclin D1 gene amplification (36), were obtained from the American Type Culture Collection (ATCC, Rockville, MD). The cultured cells were grown to approximately 70% confluence under standard cell culture conditions in an incubator supplemented with 5% CO2. The cells were harvested using 0.05% trypsin-EDTA (Sigma-Aldrich, St. Louis, MO) and were centrifuged at 1000 rpm for 5 minutes to form pellets. The cell pellets were placed in 3% bacto-agar (Difco, Detroit, MI), fixed in 10% buffered formalin, and then embedded in paraffin. These cell lines were used as controls for cyclin D1 gene amplification.
Microdissection and DNA Extraction
Ten-micrometer-thick tissue sections were cut from the paraffin blocks (tumor tissue and cell blocks) and placed on glass slides using standard precautions to avoid cross-contamination between cases (15). These precautions included cutting one case at a time, changing microtome blades between cases, floating the section in its own water bath, and cleaning the work area of the microtome with ethanol between blocks. The sections were dried at 37° C overnight, deparaffinized in xylene, rehydrated in graded ethanols, and then stained with hematoxylin for 30 seconds. Lesional tissue was microdissected using a scalpel tip under a dissecting light microscope (Laborlux 6000: Leica, Toronto, Canada). Areas of fibrosis, necrosis, and hemorrhage were excluded from microdissection. The tissue was then transferred to a microfuge tube and incubated overnight at 55° C in 50 μL of digestion buffer containing 50 mm Tris-HCl (pH 8.5), 10 mm EDTA, 0.5% Tween-20, and 0.5 mg/mL Proteinase K (Sigma-Aldrich, St. Louis, MO). After incubation, the samples were heated at 95° C for 15 minutes to inactivate the Proteinase K, and then they were stored at 4° C until used for polymerase chain reaction (PCR).
Semiquantitative Differential Polymerase Chain Reaction
An aliquot of the extracted DNA was used for the differential PCR with primers specific for a 152-bp fragment of the cyclin D1 (CD1) gene and a 112-bp fragment of the dopamine receptor (DR) gene, the latter serving as an internal control. Table 1 shows the primers used (37, 38). Differential PCR was performed in a total volume of 15 μL, which contained either 1 or 2 μL of template DNA, PCR buffer (20 mm Tris-HCl [pH 8.4], 50 mm KCl [Life Technologies, Burlington, Canada]), 1.5 mm MgCl2, 100 μm of each dNTP, 1 U of Platinum Taq DNA Polymerase (Life Technologies), 0.4 μm of cyclin D1 primers, and 0.3 μm of dopamine receptor primers. The PCR amplification conditions consisted of 5 minutes of denaturation at 94° C, followed by 30 cycles of the following: denaturation for 1 minute at 94° C, annealing for 1 minute at 53° C, elongation for 1 minute at 72° C. Amplification ended with a final extension for 5 minutes at 72° C. Each sample was analyzed at least twice in separate PCRs. DNA extracted from the paraffin-embedded cell lines served as positive (ZR-75-1) and negative (MDA-MB-231) controls for cyclin D1 amplification.
Analysis of the PCR Products
The PCR products were separated by electrophoresis on a 10% polyacrylamide gel. The gels were stained with ethidium bromide and visualized using a UVP bioimaging system (UVP, Upland, CA). The ratio between the densities of the cyclin D1 PCR product and the dopamine receptor PCR product was measured in triplicate from pictures of the stained gels using the UVP system and LabWorks Image Acquisition and Analysis software 4.0 (UVP, Upland, CA). A mean ratio of the CD1 to DR density was calculated for each sample. Cyclin D1 gene amplification was defined as 3 standard deviations (SD) above the mean ratio of the negative control, MDA-MB-231 (CD1/DR ratio of ≥0.9, n = 20).
Immunohistochemistry
Four-micrometer sections were cut from paraffin blocks and mounted on snowcoat X-tra glass slides (Surgipath, Winnipeg, Canada). Sections were dewaxed in xylene, rehydrated in ethanol, and had endogenous peroxidase activity blocked using 3% hydrogen peroxide. The immunoreactivity of the antigen was enhanced by microwaving the sections for 18 minutes in 10 mm citrate buffer, pH 6.0. The sections were incubated with a blocking reagent (Ultra Streptavidin Detection System, Signet Laboratories, Dedham, MA; 1:5 dilution) for 10 minutes, then incubated for 1 hour at room temperature with antibodies reactive with one of the following: cyclin D1 protein (monoclonal, dilution 1:50, clone DCS-6; DAKO Diagnostics, Mississauga, Canada), cyclin D3 protein (monoclonal, dilution 1:20, clone DCS-22; Novocastra, Newcastle-upon-Tyne, UK), cyclin B1 protein (monoclonal, dilution 1:20, clone 7A9; Novocastra), or Ki-67 protein (monoclonal, dilution 1:600, clone Mib1; Beckman Coulter, Mississauga, Canada). After washing, the sections were incubated for 30 minutes at room temperature with biotinylated horse-antimouse immunoglobulin G (dilution 1:200; Vector Laboratories, Burlington, Canada), followed by avidin-biotin peroxidase (ABC) complex (Vectastain Elite ABC Kit, dilution 1:2; Vector Laboratories). Immunoreactivity was visualized with 3,3′-diaminobenzidine (Sigma-Aldrich, St. Louis, MO), and sections were counterstained briefly with Mayer’s hematoxylin. The negative control consisted of replacing the primary antibody with nonimmune mouse serum (DAKO). Tissue sections from a formalin-fixed, paraffin-embedded lymph node known to express all the different proteins were used as positive controls. For cyclin D1, D3, and MIB-1 antibodies, nuclear staining indicated a positive reaction. Cytoplasmic staining was considered nonspecific and interpreted as negative. Both nuclear and cytoplasmic staining for cyclin B1 was considered indicative of a positive reaction. The percentages of giant cells and mononuclear cells showing immunopositivity with each antibody were semiquantitatively assessed by two of the authors (AK, RK) as follows: 1+, 0–5% of cells positive; 2+, 6–50% of cells positive; 3+, >50% of cells positive. If there was a discrepancy of >10% between reviewers, the case was reviewed on a multiheaded microscope, and the percentage positivity was reevaluated. Immunopositivity in >5% of cells was considered indicative of overexpression.
Statistical Analysis
The chi-square test was used to compare the clinical characteristics of the patient population. A P value of <.05 was considered statistically significant.
RESULTS
Clinical and Pathological Findings
Forty-six samples from 41 patients coded as giant cell tumor of bone were retrieved from the files of Mount Sinai Hospital. Fourteen cases were excluded from the study, because there was insufficient or inadequate material for analysis (6 cases), because the tissue was decalcified (4 cases), or because the sample was from a recurrent lesion (4 cases). This resulted in a total of 32 cases of giant cell tumor that met the requirements for inclusion in the study. The average age for these patients was 33.9 ± 14.5 years (mean ± SD), with a median age of 28 years. There were 15 females (47%) and 17 males (53%). There was no statistically significant age difference between the two genders (P = .196). The distal femur was the commonest site of involvement. Table 2 summarizes the clinical characteristics of the patients at the time of presentation as well as the location of their tumors.
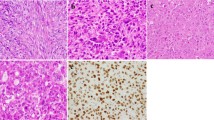
Histologically, all lesions were composed of ovoid to spindle-shaped mononuclear cells interspersed between variable numbers of multinucleated giant cells (Fig. 1). Extravasated red blood cells were seen in all cases. The giant cells were evenly distributed throughout the lesion in 27 cases (84%) and were clustered in areas of hemorrhage or extravasated red blood cells in the remaining 5 cases (16%). Fibrosis, necrosis, and infiltration by foamy histiocytes were also common features. Aneurysmal bone cyst coexisted with giant cell tumor in 8 (25%) cases.

Photomicrograph of a typical case of giant cell tumor showing multinucleated giant cells interspersed among mononuclear cells. (H&E; original magnification, 200 ×).
Semiquantification of Cyclin D1 Amplification
To evaluate the sensitivity and reproducibility of the differential PCR assay in detecting cyclin D1 gene amplification, two human breast carcinoma-derived cell lines, ZR-75-1 and MDA-MB-231, with known cyclin D1 gene copy numbers were used. DNA extracted from the formalin-fixed, paraffin-embedded cell lines was evaluated in 20 separate differential PCRs, and the mean cyclin D1/dopamine receptor ratio (CD1/DR) ± SD was calculated for each cell line. For ZR-75-1, which has a twofold to fivefold cyclin D1 gene amplification, the ratio was 1.41 ± 0.15 (mean ± SD; 95% CI: 1.35–1.47), and for MDA-MB-231, which has no cyclin D1 gene amplification, it was 0.61 ± 0.09 (mean ± SD; 95% CI: 0.55–0.67). The CD1/DR ratio of the negative control never reached the value of 1.35, which was the lower limit of the 95% CI of the positive control’s ratio. Therefore, differential PCR using dopamine receptor as a reference gene is suitable for the detection of cyclin D1 gene amplification using formalin-fixed, paraffin-embedded material and is sufficiently sensitive to detect a twofold to fivefold amplification. On the basis of these analyses, a value of ≥0.9 was considered indicative of cyclin D1 gene amplification. This value is 3 SDs above the mean CD1/DR ratio of the negative control.
To semiquantitatively estimate the level of cyclin D1 gene amplification in giant cell tumor, genomic DNA was extracted and evaluated by differential PCR in 31/32 cases. In one case, DNA could not be extracted. The mean CD1/DR ratio in the tumors ranged from 0.32 to 1.00 (Table 3). There were 19 (61%) cases with a CD1/DR ratio of ≥0.9, indicative of cyclin D1 gene amplification. No case attained a ratio of 1.35 (the lower limit of the 95% CI of the positive control), suggesting that there was a very low level of cyclin D1 gene amplification. Gene amplification occurred in 9 (70%) of the 13 cases that occurred in the distal femur. Three (75%) of the four cases that presented in the distal radius and had genomic DNA available for analysis showed amplification. However, cases presenting at other sites were associated with the presence of low levels of cyclin D1 gene amplification. There seems to be no correlation between the site and the presence of amplification. A representative polyacrylamide gel showing PCR products from five cases of giant cell tumor, including positive and negative controls, is shown in Figure 2.

Ethidium bromide-stained polyacrylamide gel showing PCR products from five cases of giant cell tumor. The PCR products for cyclin D1 (CD1) and dopamine receptor (DR) are indicated, as well as the CD1/DR ratio for each case and for the controls. Cases 4 and 5 showed low-level cyclin D1 gene amplification, whereas Cases 1 to 3 did not show amplification. ZR-75-1 cell line, which shows twofold to fivefold cyclin D1 amplification, was used as a positive (+) control, and MDA-MB-231 cell line, which shows no cyclin D1 amplification, was used as the negative (−) control.
Immunohistochemical Findings
Immunohistochemical staining was evaluated without knowledge of the gene amplification results. The percentages of giant cells and mononuclear cells showing immunopositivity for cyclins D1, D3, and B1 and for Ki-67 (MIB-1) are summarized in Tables 3 and 4. The pattern and distribution of immunopositivity was very characteristic for the different antibodies used. All cases showed cyclin D1 overexpression. Cyclin D1 was seen predominantly in the giant cells. When a giant cell was positive, all the nuclei showed immunoreactivity (Fig. 3). Twenty-three cases (72%) showed cyclin D1 positivity in >50% of the giant cells, and the remaining 9 cases showed staining in 10–50% of the giant cells (Table 3). Cyclin D1 was also seen in mononuclear cells; however, only five cases (16%) showed staining in >5% of these cells (Table 4). Cyclin D3 staining was present in all cases, but 28 cases (88%) showed cyclin D3 overexpression. Cyclin D3 distribution was similar to cyclin D1, as staining was seen predominantly in the giant cells (Fig. 4).

Immunohistochemical staining with antibody reactive with cyclin D1 shows immunopositivity predominantly in the nuclei of the giant cells. Some mononuclear cells are also positive (arrow). (Immunoperoxidase with hematoxylin counterstain; original magnification, 200 ×).

Cyclin D3 immunoreactivity is present predominantly in the giant cells, with occasional mononuclear cells showing positivity. This distribution was similar to that seen for cyclin D1. (Immunoperoxidase with hematoxylin counterstain; original magnification, 200 ×).
Cyclin B1 protein expression was seen in all cases. However, in contrast to cyclin D1 and D3, cyclin B1 staining was restricted to the mononuclear cells. Both nuclear and cytoplasmic staining was seen. In all cases, the giant cells were negative (Fig. 5). Fourteen of 32 cases (44%) showed cyclin B1 immunopositivity in >5% of the mononuclear cells. Of these 14 cases, 2 showed both cyclin D1 and cyclin B1 overexpression.

Immunohistochemical staining for cyclin B1 antibody shows immunoreactivity exclusively in the mononuclear cells. There is nuclear (arrow) and/or cytoplasmic (arrowhead) staining. No staining is seen in the giant cells. (Immunoperoxidase with hematoxylin counterstain; original magnification, 400 ×).
Ki-67 (MIB-1) staining was present in all cases and was restricted to the nuclei of the mononuclear cells (Fig. 6). No staining was seen in the giant cells. MIB-1 staining occurred in 10 to 50% of the mononuclear cells (Table 4).

Ki-67 is expressed exclusively in the nuclei of the mononuclear cells. The giant cells are negative. (Immunoperoxidase with hematoxylin counterstain; original magnification, 200 ×).
Correlation of Gene Amplification and Protein Overexpression
All cases with cyclin D1 gene amplification showed protein overexpression. However, cyclin D1 protein overexpression occurred in the absence of gene amplification in 12 of 31 cases (39%) of giant cell tumor examined.
DISCUSSION
In this report, we demonstrated the presence of low-level cyclin D1 gene amplification in 19/31 cases of giant cell tumor of bone using a differential PCR assay. Immunohistochemical staining revealed that cyclin D1 protein overexpression was present in all cases. Staining was seen in both the giant cells and mononuclear cells, although more giant cells showed protein expression than mononuclear cells. Cyclin D3 protein expression showed a similar distribution. Cyclin D1 and/or D3 staining in the giant cells was never associated with the expression of either cyclin B1 or the proliferation marker MIB-1 because staining for the latter two proteins was restricted to the mononuclear cells.
The significance of the low level of cyclin D1 gene amplification in giant cell tumor is not clear. The differential PCR technique used to assess cyclin D1 amplification in this study has been used by others for the semiquantitative analysis of gene amplification in DNA extracted from formalin-fixed, paraffin-embedded tissue (15, 16, 39), so this low-level amplification is unlikely to be an artifact of the technique. Dopamine receptor was used as the reference gene because it is located on the same arm of chromosome 11 as the cyclin D1 gene. Therefore, this assay of gene amplification will be unaffected by aneuploidy associated with extra copies of chromosome 11 or multiple copies of its long arm (37). However, we cannot rule out the possibility of amplification of the dopamine receptor gene, which would result in underestimation of the number of cases with cyclin D1 gene amplification. This is considered very unlikely because a PubMed search indicates no reports of dopamine receptor amplification. Low-level cyclin D1 gene amplification has been observed in other human tumors and been implicated in their pathogenesis (40, 41, 42). The low level of cyclin D1 gene amplification detected in giant cell tumor in this study might be the result of dilution by cells that do not have this alteration. For example, most of the mononuclear cells did not show cyclin D1 protein expression by immunohistochemistry, suggesting that they may not have gene amplification. To address this issue, five cases of giant cell tumor were dissected using a laser capture microdissection technique to obtain a giant cell-enriched population. The CD1/DR ratios obtained from the laser- and scalpel-microdissected tissue were comparable (data not shown), suggesting that the mononuclear cells were unlikely to be responsible for the low values of gene amplification detected by differential PCR. However, further study, such as real-time quantitative PCR on a pure population of giant cells or quantitative fluorescence in situ hybridization, might help to determine more accurately the levels of gene amplification in these tumors.
Cyclin D1 protein overexpression was detected in all giant cell tumor cases. Although all cases having gene amplification had protein overexpression, some cases demonstrated protein overexpression in the absence of gene amplification. This observation has been reported by others (15, 16) and suggests that mechanisms other than gene amplification may play a role in cyclin D1 protein overexpression. For example, chromosomal rearrangement, such as t(11;14) translocation seen in mantle cell lymphomas, leads to cyclin D1 gene activation, increased cyclin D1 mRNA, and consequently, to high levels of the protein (43). Other possibilities include alterations of gene transcription, mRNA translation, or protein degradation. Continuous mitogen stimulation or growth factor activation of receptor tyrosine kinases lead to increased transcription of cyclin D genes and decreased turnover of the protein (44, 45). Cyclin D availability can be regulated at the translational level through the phosphatidylinositol 3-kinase (PI 3-kinase) pathway, which is involved in the G1-to-S phase progression in response to a variety of growth factors (46). Dysregulation of this pathway can result in cyclin D1 protein overexpression in the absence of gene amplification. In addition, events affecting cyclin D1 turnover might play a role in the stabilization of the protein and the ability to detect it immunohistochemically. Cyclin D1 is degraded via the ubiquitin/proteasome pathway, and this degradation is dependent on phosphorylation of specific residues by glycogen synthase kinase-3β (GSK-3β) (47). Inhibition of this phosphorylation prevents rapid cyclin D1 degradation (45). Finally, an alternative explanation for the lack of correlation between cyclin D1 gene amplification and protein levels might be methodological issues related to the PCR, the sensitivity of the antibody, or variations in the length of formalin fixation of the tissue.
This study demonstrated proliferative activity in giant cell tumor that is in keeping with that found by other studies. At least five reports have documented the presence of proliferative activity in giant cell tumor (29, 30, 31, 32, 33), but these have focused on the assessment of Ki-67 (MIB-1) expression. The expression of cyclin D1, D3, B1, and Ki-67 in the mononuclear cells is consistent with proliferation of these cells. In contrast, overexpression of cyclin D1 and D3 in the giant cells is not accompanied by Ki-67 or cyclin B1 expression. There are two possible explanations for the aberrant expression of cyclins D1 and D3 in the giant cells of giant cell tumor. First, this may reflect the presence of deregulated cell cycle control in giant cell tumor. It is possible that the giant cells are arrested at the G1/S transition and do not enter the M phase (as evidenced by the absence of cyclin B1 expression). However, if this is the mechanism, it is not clear why there is no Ki-67 expression detected in the giant cells, as this protein is expressed in all cycling cells. Alternatively, it may be that cyclin D1 is playing a role in giant cell formation. The observation that the percentage of giant cells showing cyclin D1 immunopositivity varies within the tumors (range, 10–90%) does not rule this out because this might be due to differences in the length of formalin fixation of the tissue, in tumor sampling, or in the stage of giant cell maturation. Cyclin D1 protein overexpression has been shown to be associated with giant cell formation, multinucleation, and increased ploidy in different cell models. Cyclin D1 overexpression in transgenic mice was shown to result in sustained cardiomyocyte DNA synthesis in adult hearts and in a multinucleated phenotype (48). Cyclin D1 transcripts were found in giant mouse trophoblast cells but not in diploid cells. Cyclin D1 expression was evident early in the development of the giant trophoblast cells, whereas cyclin B1 protein was not detected (49). Overexpression of cyclin D3, and to a lesser extent, cyclin D1, is thought to play a key role in the endomitotic cell cycle of megakaryocytes (50), and antisense oligonucleotides designed to suppress cyclin D3 expression were shown to abrogate endomitosis and megakaryocyte development (51). Although the possibility of endomitosis as the mechanism underlying the formation of giant cells in giant cell tumor has been suggested previously (52), this could not be confirmed by the present study, mainly because of the absence of Ki-67 expression in these cells, which should occur in cells undergoing endomitosis (53). The consistent overexpression of the D cyclins studied and the absence of cyclin B1 staining in the giant cells, both findings described in the endomitotic cell cycle of megakaryocytes, suggests that endomitosis may be occurring in the giant cells. Alternatively, it is possible that overexpression of cyclin D1 and D3 plays a role in the formation of the giant cells in giant cell tumor through other mechanisms that are not clearly understood at the present time.
Giant cell tumor of bone is a locally aggressive tumor whose clinical behavior is difficult to predict based on its microscopic appearance alone (31). Several studies directed toward determining prognostic indicators for these tumors have not identified any. Although we did not attempt to evaluate the prognostic significance of the expression of the different cyclins in giant cell tumor, the detection of cyclins D1 and D3 overexpression in all of our cases makes it unlikely that the D cyclins could be used to predict the clinical behavior of this tumor. Nevertheless, our results suggest that alterations in cyclin D1 and/or D3 might play a role in the pathogenesis and the formation of giant cells in giant cell tumor. However, additional studies are needed to further clarify the functional role of these cell cycle regulatory molecules in giant cell tumor. Similarly, study of lesions with similar histologic features, such as central giant cell granuloma of the jaws, might shed more light on such a role. A study comparing central giant cell granuloma and giant cell tumor is currently ongoing in our laboratory.
References
Sherr CJ, Roberts JM . CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13: 1501–1512.
Mittnacht S . Control of pRB phosphorylation. Curr Opin Genet Dev 1998; 8: 21–27.
Sherr CJ . Cancer cell cycles. Science 1996; 274: 1672–1677.
Ekholm SV, Reed SI . Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol 2000; 12: 676–684.
Lukas J, Muller H, Bartkova J, et al. DNA tumor virus oncoproteins and retinoblastoma gene mutations share the ability to relieve the cell’s requirement for cyclin D1 function in G1. J Cell Biol 1994; 125: 625–638.
Resnitzky D, Gossen M, Bujard H, Reed SI . Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol Cell Biol 1994; 14: 1669–1679.
Liu SC, Klein-Szanto AJ . Markers of proliferation in normal and leukoplakic oral epithelia. Oral Oncol 2000; 36: 145–151.
Bartek J, Bartkova J, Lukas J . The retinoblastoma protein pathway in cell cycle control and cancer. Exp Cell Res 1997; 237: 1–6.
Lukas J, Aagaard L, Strauss M, Bartek J . Oncogenic aberrations of p16INK4/CDKN2 and cyclin D1 cooperate to deregulate G1 control. Cancer Res 1995; 55: 4818–4823.
Bodrug SE, Warner BJ, Bath ML, et al. Cyclin D1 transgene impedes lymphocyte maturation and collaborates in lymphomagenesis with the myc gene. EMBO J 1994; 13: 2124–2130.
Hinds PW, Dowdy SF, Eaton EN, Arnold A, Weinberg RA . Function of a human cyclin gene as an oncogene. Proc Natl Acad Sci U S A 1994; 91: 709–713.
Lee RJ, Albanese C, Fu M, et al. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol 2000; 20: 672–683.
Wang TC, Cardiff RD, Zukerberg L, et al. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature 1994; 369: 669–671.
Donnellan R, Chetty R . Cyclin D1 and human neoplasia. Mol Pathol 1998; 51: 1–7.
Zhu XL, Hartwick W, Rohan T, Kandel R . Cyclin D1 gene amplification and protein expression in benign breast disease and breast carcinoma. Mod Pathol 1998; 11: 1082–1088.
Rousseau A, Lim MS, Lin Z, Jordan RC . Frequent cyclin D1 gene amplification and protein overexpression in oral epithelial dysplasias. Oral Oncol 2001; 37: 268–275.
Capaccio P, Pruneri G, Carboni N, et al. Cyclin D1 protein expression is related to clinical progression in laryngeal squamous cell carcinomas. J Laryngol Otol 1997; 111: 622–626.
Jackman M, Firth M, Pines J . Human cyclins B1 and B2 are localized to strikingly different structures: B1 to microtubules, B2 primarily to the Golgi apparatus. EMBO J 1995; 14: 1646–1654.
Brandeis M, Rosewell I, Carrington M, et al. Cyclin B2-null mice develop normally and are fertile whereas cyclin B1- null mice die in utero. Proc Natl Acad Sci U S A 1998; 95: 4344–4349.
Yang J, Bardes ES, Moore JD, et al. Control of cyclin B1 localization through regulated binding of the nuclear export factor CRM1. Genes Dev 1998; 12: 2131–2143.
Soria JC, Jang SJ, Khuri FR, et al. Overexpression of cyclin B1 in early-stage non-small cell lung cancer and its clinical implication. Cancer Res 2000; 60: 4000–4004.
Kawamoto H, Koizumi H, Uchikoshi T . Expression of the G2-M checkpoint regulators cyclin B1 and cdc2 in nonmalignant and malignant human breast lesions: immunocytochemical and quantitative image analyses. Am J Pathol 1997; 150: 15–23.
Wang A, Yoshimi N, Ino N, Tanaka T, Mori H . Overexpression of cyclin B1 in human colorectal cancers. J Cancer Res Clin Oncol 1997; 123: 124–127.
Mashal RD, Lester S, Corless C, et al. Expression of cell cycle-regulated proteins in prostate cancer. Cancer Res 1996; 56: 4159–4163.
Kushner J, Bradley G, Young B, Jordan RC . Aberrant expression of cyclin A and cyclin B1 proteins in oral carcinoma. J Oral Pathol Med 1999; 28: 77–81.
Murakami H, Furihata M, Ohtsuki Y, Ogoshi S . Determination of the prognostic significance of cyclin B1 overexpression in patients with esophageal squamous cell carcinoma. Virchows Arch 1999; 434: 153–158.
Hassan KA, El-Naggar AK, Soria JC, et al. Clinical significance of cyclin B1 protein expression in squamous cell carcinoma of the tongue. Clin Cancer Res 2001; 7: 2458–2462.
Dorfman HD, Bogdan C, editors. Bone tumors. 1st ed. Toronto: Mosby, Inc.; 1998.
Masui F, Ushigome S, Fujii K . Giant cell tumor of bone: a clinicopathologic study of prognostic factors. Pathol Int 1998; 48: 723–729.
Masui F, Ushigome S, Fujii K . Giant cell tumor of bone: an immunohistochemical comparative study. Pathol Int 1998; 48: 355–361.
Sulh MA, Greco MA, Jiang T, et al. Proliferation index and vascular density of giant cell tumors of bone: are they prognostic markers? Cancer 1996; 77: 2044–2051.
Scotlandi K, Serra M, Manara MC, et al. Clinical relevance of Ki-67 expression in bone tumors. Cancer 1995; 75: 806–814.
Antal I, Sapi Z, Szendroi M . The prognostic significance of DNA cytophotometry and proliferation index (Ki-67) in giant cell tumors of bone. Int Orthop 1999; 23: 315–319.
Bartkova J, Lukas J, Muller H, et al. Cyclin D1 protein expression and function in human breast cancer. Int J Cancer 1994; 57: 353–361.
Lammie GA, Fantl V, Smith R, et al. D11S287, a putative oncogene on chromosome 11q13, is amplified and expressed in squamous cell and mammary carcinomas and linked to BCL-1. Oncogene 1991; 6: 439–444.
Buckley MF, Sweeney KJ, Hamilton JA, et al. Expression and amplification of cyclin genes in human breast cancer. Oncogene 1993; 8: 2127–2133.
Gramlich TL, Fritsch CR, Maurer D, Eberle M, Gansler TS . Differential polymerase chain reaction assay of cyclin D1 gene amplification in esophageal carcinoma. Diagn Mol Pathol 1994; 3: 255–259.
Suzuki H, Moriya J, Nakahata A, et al. Cyclin D1 gene amplification in esophageal carcinosarcoma shown by differential polymerase chain reaction. Hum Pathol 1998; 29: 662–667.
Neubauer A, Neubauer B, He M, et al. Analysis of gene amplification in archival tissue by differential polymerase chain reaction. Oncogene 1992; 7: 1019–1025.
Zukerberg LR, Yang WI, Gadd M, et al. Cyclin D1 (PRAD1) protein expression in breast cancer: approximately one-third of infiltrating mammary carcinomas show overexpression of the cyclin D1 oncogene. Mod Pathol 1995; 8: 560–567.
Frierson HF Jr, Gaffey MJ, Zukerberg LR, Arnold A, Williams ME . Immunohistochemical detection and gene amplification of cyclin D1 in mammary infiltrating ductal carcinoma. Mod Pathol 1996; 9: 725–730.
Gaffey MJ, Iezzoni JC, Meredith SD, et al. Cyclin D1 (PRAD1, CCND1) and glutathione-S-transferase pi gene expression in head and neck squamous cell carcinoma. Hum Pathol 1995; 26: 1221–1226.
Sola B, Salaun V, Ballet JJ, Troussard X . Transcriptional and post-transcriptional mechanisms induce cyclin-D1 over-expression in B-chronic lymphoproliferative disorders. Int J Cancer 1999; 83: 230–234.
Daksis JI, Lu RY, Facchini LM, Marhin WW, Penn LJ . Myc induces cyclin D1 expression in the absence of de novo protein synthesis and links mitogen-stimulated signal transduction to the cell cycle. Oncogene 1994; 9: 3635–3645.
Diehl JA, Zindy F, Sherr CJ . Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev 1997; 11: 957–972.
Takuwa N, Fukui Y, Takuwa Y . Cyclin D1 expression mediated by phosphatidylinositol 3-kinase through mTOR-p70(S6K)-independent signaling in growth factor-stimulated NIH 3T3 fibroblasts. Mol Cell Biol 1999; 19: 1346–1358.
Diehl JA, Cheng M, Roussel MF, Sherr CJ . Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998; 12: 3499–3511.
Soonpaa MH, Koh GY, Pajak L, et al. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest 1997; 99: 2644–2654.
Palazon LS, Davies TJ, Gardner RL . Translational inhibition of cyclin B1 and appearance of cyclin D1 very early in the differentiation of mouse trophoblast giant cells. Mol Hum Reprod 1998; 4: 1013–1020.
Sun S, Zimmet JM, Toselli P, et al. Overexpression of cyclin D1 moderately increases ploidy in megakaryocytes. Haematologica 2001; 86: 17–23.
Wang Z, Zhang Y, Kamen D, Lees E, Ravid K . Cyclin D3 is essential for megakaryocytopoiesis. Blood 1995; 86: 3783–3788.
Grano M, Colucci S, De Bellis M, et al. New model for bone resorption study in vitro: human osteoclast-like cells from giant cell tumors of bone. J Bone Miner Res 1994; 9: 1013–1020.
Pellegrini W, Facchetti F, Marocolo D, et al. Assessment of cell proliferation in normal and pathological bone marrow biopsies: a study using double sequential immunophenotyping on paraffin sections. Histopathology 1995; 27: 397–405.
Acknowledgements
Supported by a grant from the Canadian Institute for Health Research (IHRT).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kauzman, A., Li, S., Bradley, G. et al. Cyclin Alterations in Giant Cell Tumor of Bone. Mod Pathol 16, 210–218 (2003). https://doi.org/10.1097/01.MP.0000057235.65327.40
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000057235.65327.40
Keywords
This article is cited by
-
Riesenzelltumor des Knochens
Der Pathologe (2020)
-
Characterization of Three Novel H3F3A-mutated Giant Cell Tumor Cell Lines and Targeting of Their Wee1 Pathway
Scientific Reports (2019)
-
Central giant cell lesion of the jaws: study of CCND1 gene amplification and p16INK4a protein levels
Journal of Molecular Histology (2013)
-
p53 mutations may be involved in malignant transformation of giant cell tumor of bone through interaction with GPX1
Virchows Archiv (2013)
-
Centrosome abnormalities in giant cell tumour of bone: possible association with chromosomal instability
Modern Pathology (2010)
