
Abstract
Tissue microarrays allow high throughput molecular profiling of diagnostic or predictive markers in cancer specimens and rapid validation of novel potential candidates identified from genomic and proteomic analyses in a large number of tumor samples. To validate the use of tissue microarray technology for all the main biomarkers routinely used to decide breast cancer prognostication and postsurgical adjuvant therapy, we constructed a tissue microarray from 97 breast tumors, with a single 0.6 mm core per specimen. Immunostaining of tissue microarray sections and conventional full sections of each tumor were performed using well-characterized prognostic markers (estrogen receptor ER, progesterone receptor PR and c-erbB2). The full section versus tissue microarray concordance for these stains was 97% for ER, 98% for PR, and 97% for c-erbB2, respectively, with a strong statistical association (kappa value more than 0.90). Fluorescence in situ hybridization analysis for HER-2/neu gene amplification from the single-core tissue microarray was technically successful in about 90% (87/97) of the cases, with a concordance of 95% compared with parallel analyses with the full sections. The correlation with other pathological parameters was not significantly different between full-section and array-based results. It is concluded that the constructed tissue microarray with a single core per specimen ensures full biological representativeness to identify the associations between biomarkers and clinicopathological parameters, with no significant associated sampling bias.
Similar content being viewed by others

INTRODUCTION
Both genetic and environmental factors play a key role in the development of breast cancer, one of the commonest cancers in the world. Understanding the basis of tumor development and progression, and identifying biomarkers for assessment of prognosis and prediction of therapy outcome are integral parts of current research efforts. Traditionally, three well-characterized biomarkers—estrogen receptor or ER (1, 2, 3, 4), progesterone receptor or PR (3, 4), and c-erbB2 or HER-2/neu oncogene (5, 6, 7)—have been used in the clinical analysis of breast cancer by immunohistochemistry and fluorescence in situ hybridization (FISH), both applied to full sections of formalin-fixed, paraffin-embedded tumor tissues. However, this would be time-consuming and tedious when processing large numbers of tumors or when screening with multiple markers. The recently developed tissue microarray, composed from multiple donor tumors systematically aligned within a single recipient block, allows for high throughput molecular profiling of many tumor tissues simultaneously in a single experiment. Thin sections cut from such tissue microarray blocks can be used to study gene amplification and protein overexpression by DNA and RNA in situ hybridization or by immunohistochemistry, with tremendous savings in analysis time, labor and reagent costs. This technology has the potential to significantly accelerate studies seeking for the association between molecular changes and clinical endpoints (8, 9) and the validation of novel biomarkers (10). The high speed of arraying, the lack of any significant damage to the donor blocks, and the uniform treatment and assessment of regularly arrayed tumors representing different tumor stages, histological grades, and histological subtypes are the most significant advantages of the tissue microarray technology. Use of tissue microarrays will greatly accelerate the transition of basic research findings to clinical applications.
One of the limitations of the tissue microarray technology is that the “punched” minute samples from donor tissues may not always be representative of the entire tumors with their well-established heterogeneity. Therefore, “punching” from multiple sites from each original block was recommended to reduce the sampling problem (11, 12, 13). However, it was reported in a recent study that a single “punch” uncovered most of the information on ER expression in breast cancer, and that tumor heterogeneity did not negatively influence the predictive power of tissue microarray results (9). The associations between molecular changes and clinical endpoints detected on tissue microarrays based on the single core specimen per tumor suggest that biomarkers with prognostic significance are more likely to be expressed in a relatively homogeneous manner in the tumor. The more homogeneously a marker of interest is expressed in the tumor and the more histologically homogeneous the tumor is, the more likely the tissue microarray data will reproduce the findings from the full sections. Sampling a typical and representative region of the tumor is certainly the key step in tissue microarray construction. Therefore, careful selection of representative regions of tumors and precise “punching” from donor tissues can reproduce sufficient information in cell populations.
In the present study, we investigated the efficiency and representativeness of immunohistochemistry and FISH analyses in our constructed tissue microarrays of breast cancer. We constructed a tissue microarray with a single 0.6 mm core biopsy per specimen and applied the most commonly used diagnostic and prognostic markers, including monoclonal antibodies against ER, PR and c-erbB2 by immunohistochemistry and the locus-specific HER-2/neu probe by FISH. We defined the concordance of data from both tissue microarray and full sections and the impact of data discrepancy between the two methods with regard to the correlation with other pathological parameters.
MATERIALS AND METHODS
Patients
Ninety-seven breast cancer samples from the National University Hospital of Singapore were included in this study. The age ranged from 32 to 86 years, with an average of 53 years. The tumors included invasive ductal carcinoma (n = 84), ductal carcinoma in situ (n = 7), medullary carcinoma (n = 3) and mucinous carcinoma (n = 3). Forty-six cases had metastases to lymph nodes while 51 were node-free. Among the 97 tumors, 11.3% (n = 11) had a histologic grade 1, 39.2% (n = 38) a grade 2, and 49.5% (n = 48) a grade 3.
Tissue Microarray Construction
A total of 194 formalin-fixed, paraffin-embedded breast tissue samples (97 tumors and 97 normal) were used for tissue microarray construction, as described by Kononen et al. (9). Ninety-seven tumors, with 5 normal tissues as internal negative controls, were arrayed in one paraffin block and 97 normal tissues, in another. Before arraying, 4-μm sections from each tissue block were stained with Hematoxylin and Eosin (H&E) and a morphologically representative area of tumor was defined by the pathologists. Tissue cylinders with a diameter of 0.6 mm were then punched from the targeted tumor area of each donor tissue block and deposited into a recipient block using a tissue arraying instrument (Beecher Instruments®, Silver Spring, MD). After construction of the array block, multiple consecutive 4-μm sections were cut until all the 97 tissue samples were represented on a single section. One such section was placed on a microscopic slide and H&E-stained for histological verification of the adequacy of the arrayed tumor tissues. The consecutive sections were separately placed on charged polylysine-coated slides for immunohistochemistry and FISH analyses. Sections from the second tissue microarray of normal samples were prepared by the same process and served as controls.
Immunohistochemistry
Standard indirect immunoperoxidase procedures were used for immuno-histochemistry. Sections from formalin-fixed, paraffin-embedded tissues were deparaffinized, treated with 3% hydrogen peroxide in phosphate-buffered saline and pretreated at 95°C for 10 minutes in 10 μmol/L citrate buffer (pH 6.0). Three monoclonal antibodies were used for detection of ER (ER1D5, 1:1000), PR (NCL-PGR, 1A6, 1:1000) and c-erbB2 (HercepTest™ K5204, 1:500), all from DAKO®. Diaminobenzidine was used as the chromogen and hematoxylin as the nuclear counterstain. The same scoring criteria were applied to the tissue microarray and the large sections. All slides were independently analyzed by the pathologists (MST and TCP) and the principal researcher (ZDH). Tumors were considered positive for ER and PR if more than 10% of tumor cells showed an unequivocal nuclear staining. Tumors exhibiting definite cytoplasmic membrane staining (grade 2+ and 3+) were considered to be positive for c-erbB2 expression.
Fluorescence In Situ Hybridization (FISH)
The tissue array sections were treated according to the Paraffin Pretreatment Reagent Kit (Vysis®) protocol, before hybridization. FISH was performed with a locus-specific Spectrum Orange-labeled HER-2/neu probe and a Spectrum Green-labeled chromosome 17 centromeric probe (CEP-17) as an internal control reference probe. Hybridization and posthybridization washes were carried out according to the LSI HER-2/neu kit (Vysis®) procedure. Briefly, the deparaffinized slides were immersed in 0.2 mol/L HCl for 20 minutes and after washing with water and Wash Solution, were incubated in 1 mol/L sodium thiocyanate solution at 80°C for 30 minutes. The pretreated slides were further treated with protease solution (0.5 mg/mL in 0.9% NaCl, Vysis®) for 20 minutes at 37°C, fixed in 10% buffered formalin for 10 minutes, air-dried and denatured for 5 minutes. The probe mixture was applied to each slide, with minimal delay. After hybridization at 37°C overnight, the sections were washed and counterstained with 0.2 μmol/L DAPI (4,6-diamidino-2-phenylindole). The fluorescent signals were scored with an Olympus fluorescence microscope equipped with double-bandpass filters for simultaneous visualization of Spectrum Green and Spectrum Orange signals. The relative ratio of the orange HER-2/neu gene signals to the green chromosome 17 centromeric signals was evaluated by the cytogeneticist (ED), with a ratio of ≥2:1 arbitrarily defined as evidence for HER-2/neu gene amplification.
Statistical Analysis
The association of the results obtained with the full sections and those with the tissue microarray sections was studied using kappa statistics (14). A kappa value more than 0.5 was considered to denote a strong association between the two sets of results. Contingency table analysis and χ2 test were used to analyze the correlations of immunohistochemistry and FISH in full sections and tissue microarray with other pathological parameters. The survival curves were not described as all the cases were diagnosed in the years 2000 and 2001.
RESULTS
An overview of the results we obtained is presented in Table 1.
Efficiency of Biopsy Arraying
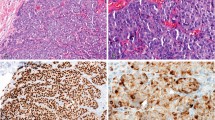
We found only 4 single-core samples missing from the H&E-stained section prepared from the tumor tissue microarray block containing 102 representative core biopsies, including 5 from normal tissues as controls. The remaining 98 samples, including the 5 normal tissues, were verified as representative core biopsies of their respective donor tumors by the pathologists (MST and TCP). The rate of non-representation for the single-core array was 4%, giving an efficiency of 96% for the 97 tumor samples. The next three consecutive sections were used for ER, PR and c-erbB2 immunohistochemistry analyses. Two other consecutive sections were prepared for H&E examination and HER-2/neu FISH analysis. Representative positive single-core results for both immunohistochemistry and FISH are shown in Figure 1.

Representative examples of ER, PR, and c-erbB2 immunohistochemistry and HER-2/neu gene amplification (FISH) in breast tumor tissue arrays. a, d, g,and j, H&E staining of four tissue samples (× 42); b, e,and h, immunostaining (× 42); c, f,and i, enlarged immunostaining of the specific area (× 200); k, a tissue microarray sample for FISH analysis (× 100); l, HER-2/neu gene amplification (× 1000): cells showing a massive increase in the number of the orange signals as compared with the green internal control signals.
Estrogen Receptor (ER) Overexpression
Ninety-three of the 97 tumor cases (91.9%) on the tissue microarray were accessible for ER analysis, with 53 positive cases (57%) showing nuclear staining in more than 10% of the tumor nuclei and 40 negative cases (40%). In comparison, for the full sections of the studied tumors, 56 cases (60.2%) were read as positive and 37 cases (39.8%) as negative. Overall, a concordance of 97% among the accessible cases and a strong statistical association (kappa value, k = 0.93) were found.
Progesterone Receptor (PR) Overexpression
Of the 93 tumor cores in the tissue microarray, 54 stained positive (58.1%). For the full sections, 56 cases (60.2%) were defined as positive. The two mismatches were negative in tissue microarray and positive in the full sections. A strong statistical association between the two methods for the PR analysis was also demonstrated (kappa value, k = 0.95).
c-erbB2 Overexpression
In the tissue microarray section, 5 tumor samples were missed during staining (technical error) and only the remaining 92 tumor cases were available for immunohistochemistry analysis. The c-erbB2 overexpression was noted in 30 cases (32.6%) while 61 cases (66.3%) were negative. In comparison, for c-erbB2 expression in the full sections, 33 of the 92 cases (34.8%) showed positivity in the full sections, with 3 cases demonstrating discrepancy, giving a non-concordance rate of 3% and a highly significant statistical association between the methods (kappa value, k = 0.92).
HER-2/neu Gene Amplification by FISH
HER-2/neu gene amplification was only scored to be present when the copy number of HER-2 gene signals exceeded that of the chromosome 17 centromeric probe (CEP-17) by at least 2-fold in 10% or more of the tumor cells. FISH analysis was successful in 87 of the 97 arrayed tumor samples (3 samples were lost and 7 others were non-informative). HER-2/neu gene amplification was detected in 20 of the 87 interpretable cases. An example of the HER-2 gene amplification by FISH in the tissue microarray section is shown in Figure 1. When matched against the corresponding FISH data from the full sections, three cases showed discrepancy. The non-concordance rate of 4.5% and the kappa value of 0.89 suggest a very good agreement of the FISH results from the two methods.
Correlation with Other Pathological Parameters
To assess the similarity (or lack thereof) of overexpression of biomarkers in relation to other pathological parameters, we independently analyzed both data sets generated from tissue microarray and full sections using the contingency table analysis and the χ2 test. From the data of the full sections, both ER and PR expressions were significantly correlated to the histological tumor grade (P < .001). The c-erbB2 expression was also strongly associated with the tumor grade (0.001 < P < .01), but in an inverse manner. A similar statistical association was noted with the tissue microarray data, including a strong relationship of ER, PR and c-erbB2 with tumor grade (P < .001). HER-2/neu amplification data from both methods showed a similar statistical association with the histological tumor grade (0.001 < P < .01). Our analysis indicated that the single-core tissue array-derived data are comparable with the data from full sections in correlating with other pathological parameters.
DISCUSSION
Tissue microarrays constructed from multiple donor tumors are a potentially useful tool for rapid and efficient analysis of large numbers of paraffin-embedded tissue (9), facilitating standardized analysis of multiple gene expression and amplification in the same tumor or the same tumor site, using the same kind of probes and standardized interpretation criteria. The main advantages of tissue microarray technology include the improved analytical capacity, the negligible damage caused to the original tissue blocks, and the precise positioning of tissue specimens on the recipient block. Usually, tissue core “punches” of 0.6 to 2.0 mm in diameter are taken from representative areas of paraffin-embedded tissues and are arrayed on a single recipient block. These punched-out cores are considered to be large enough to enable adequate histomorphology assessment and analysis of biomarker expression and amplification at the DNA, RNA and protein levels (15, 16, 17, 18). Data from three recent studies further suggest that the minute tissue samples in an array format are sufficiently representative of their donor tumors to allow authentic association studies between molecular alterations and clinical endpoints (8, 17, 19, 20). However, the tissue microarray technique is limited by the risk of non-representative sampling of the potentially heterogeneous tumors, which are often evident both at the morphological and genetic levels (21, 22, 23). Thus, the issues of what is the minimum number of “punches” per case and up to what extent the tissue microarray data can reproduce large section data are important when analyzing the clinicopathological associations. Hoos et al. (11) reported that triplicate cores offered a high rate of assessable cases and a lower rate of non-concordant readings than one or two cores, whereas Rubin et al. (13) demonstrated that four cores was optimal to represent tumor protein expression. However, this would reduce the number of tumor samples that can be assembled in a recipient block and render the process somewhat time-consuming and tedious. To maximize the efficiency of the tissue microarray technology, it is crucial to identify the minimum number of cores needed for meaningful results, to carefully select representative areas on H&E-stained full sections based on the tumor morphology and to precisely transfer the target site from the donor to the recipient blocks. We have successfully constructed a single-core tissue microarray of breast cancer wherein 93 of the 97 arrayed tumor biopsies were fully representative of the donor tissues. The current success rate in our laboratory for arraying breast cancers is greater than 95%, with loss of accessible cases limited to only 4%.
All the biomarkers analyzed in this study are the ones routinely used for breast cancer prognostication and stratification for further therapy. To identify the concordance of the immunohistochemical expression profiles of the tissue array containing a single core per specimen and the full sections, we used well-characterized antibodies against ER, PR and c-erbB2, and obtained concordance rates of 97%, 98% and 97%, respectively. Our result for PR expression was higher than the 88% concordance reported by Torhorst et al. (8). The few discrepant readings that we found in ER, PR and c-erbB2 staining changed the original abnormal results found in full sections to normal results in arrayed tissues and hence resulted in slightly lower ratios of abnormal expression patterns in tissue microarray. Full section analysis resulted in 60.2%, 60.2% and 34.8% of cases with abnormal expression of ER, PR and c-erbB2, respectively. Corresponding respective readings on the single-core tissue microarray were 57%, 58% and 32.6%. The high concordance and the strong agreement (kappa value more than 0.90) between the two methods suggest that tissue microarray can accurately reflect immunohistochemistry information currently derived from full sections.
The FISH technique has been successfully used for analysis of gene amplification by high throughput tissue microarray (16, 18, 24, 25). To validate the FISH analysis in our tissue microarray, we performed HER-2/neu gene amplification analysis and compared the data obtained with the original full-section FISH data. Seven non-interpretable samples were associated with various technical problems of FISH, such as weak hybridization signals or excessive background. The technical adequacy (87 of 97 tissues or 90%) is higher than the results (7585%) from Schraml et al. (18) and comparable with data (95.1%) from four repeats reported by Simon et al. (25). Well-defined hybridization signals were observed in 23% of the 87 accessible samples in tissue microarray, producing a 95% agreement with the data from full sections and a statistically strong association (kappa value: 0.89). However, when we used the χ2 test to compare the strength of association of the biomarker data from full sections and tissue microarray with the other pathological parameters, we did not find any significant differences between the two methods, with both showing strong associations with the other pathological parameters.
Our study included all the routinely-used biomarkers to decide patient prognosis and adjuvant therapy in breast cancer, and our data demonstrated the high reliability and comparability of tissue microarray-based immunohistochemistry and FISH using the same cut-off values for full section analysis. The biopsy of 0.6 mm diameter taken from a typical and representative region of the tumor provides a reliable and efficient system for large-scale analysis of cancer tissues on the tissue microarray platform and is useful for large-scale clinicopathological studies, conserving the molecular profile of the molecular markers that are clinically relevant.
References
Robertson JF, Bates K, Pearson D, Blarney RW, Nicholson RI . Comparison of two estrogen receptor assays in the prediction of the clinical course of patients with advanced breast cancer. J Clin Oncol 1992; 65: 727–730.
DeSombre ER, Thorpe SM, Rose C, Blough RR, Andersen KW, Rasmissen BB, et al. Prognostic usefulness of estrogen receptor immunohistochemical assays for human breast cancer. Cancer Res 1986; 46: 4256s–4264s.
Nizzoli R, Bozzetti C, Savoldi L, Manotti L, Naldi N, Camisa R, et al. Immunocytochemical assay of estrogen and progesterone receptors in fine needle aspirates from breast cancer patients. Acta Cytol 1994; 38: 933–938.
Pertschuk LP, Kim DS, Nayer K, Feldman JG, Eisenberg KB, Carter AC, et al. Immunocytochemical estrogen and progestin receptor assays in breast cancer with monoclonal antibodies: histopathologic, demographic, and biochemical correlations and relations to endocrine response and survival. Cancer 1990; 66: 1663–1670.
Tsutsui S, Ohno S, Murakami S, Hachitanda Y, Oda S . Prognostic value of c-erbB2 expression in breast cancer. J Surg Oncol 2002; 79: 216–223.
Andrulis IL, Bull SB, Blakstein ME, Sutherland D, Mak C, Sidlofsky S, et al. neu/erbB2 amplification identifies a poor-prognosis group of women with node-negative breast cancer. J Clin Oncol 1998; 16: 1340–1349.
Revillion F, Bonneterre J, Peyrat JP . ERBB2 oncogene in human breast cancer and its clinical significance. Eur J Cancer 1998; 34: 791–808.
Torhorst J, Bucher C, Kononen J, Haas P, Zuber M, Kochli OR, et al. Tissue microarrays for rapid linking of molecular changes to clinical endpoint. Am J Pathol 2001; 159: 2249–2256.
Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med 1998; 4: 844–847.
Rubin MA, Zhou M, Dhanasekaran SM, Varambally S, Barrette T, Sanda MG, et al. α-methylacyl coenzyme. A racemase as a tissue biomarker for prostate cancer. J Am Med Assoc 2002; 287: 1662–1670.
Hoos A, Urist MJ, Stojadinovic A, Mastorides S, Dudas ME, Leung DHY, et al. Validation of tissue microarrays for immunohistochemical profiling of cancer specimens using the example of human fibroblastic tumors. Am J Pathol 2001; 158: 1245–1251.
Mucci NR, Akdas G, Manely S, Rubin MA . Neuroendocrine expression in metastatic prostate cancer: evaluation of high throughput tissue microarrays to detect heterogeneous protein expression. Hum Pathol 2000; 31: 406–414.
Rubin MA, Dunn R, Strawderman M, Pienta K . Tissue microarray sampling strategy for prostate cancer biomarker analysis. Am J Surg Pathol 2002; 26: 312–319.
Altman DG . Practical statistics for medical research. London: Chapman and Hall; 1996; 404–408.
Ristimaki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, et al. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res 2002; 62: 632–635.
Bubendorf L, Kononen J, Kivisto P, Schraml P, Moch H, Gasser TC, et al. Survey of gene amplifications during prostate cancer progression by high throughput fluorescence in situ hybridization on tissue microarrays. Cancer Res 1999; 59: 803–806.
Richter J, Wagner U, Kononen J, Fijan A, Bruderer J, Schmid U, et al. High-throughput tissue microarray analysis of cyclin E gene amplification and overexpression in urinary bladder cancer. Am J Pathol 2000; 157: 787–794.
Schraml P, Konone J, Bubendorf L, Moch H, Bissig H, Nocito A, et al. Tissue microarrays for gene amplification surveys in many different tumor types. Clin Cancer Res 1999; 5: 1966–1975.
Chung GG, Provost E, Kielhorn EP, Charette LA, Smith BL, Rimm DL . Tissue microarray analysis of β-catenin in colorectal cancer shows nuclear phospho-β-catenin is associated with a better prognosis. Clin Cancer Res 2001; 7: 4013–4020.
Xie W, Mertens JC, Reiss DJ, Rimm DL, Camp RL, Haffty BG, et al. Alterations of Smad signaling in human breast carcinoma are associated with poor outcome: a tissue microarray study. Cancer Res 2002; 62: 497–505.
Allred DC, Harvey JM, Berardo M, Clasrk GM . Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol 1998; 11: 155–168.
Bergers E, van Diest PJ, Baak JP . Tumor heterogeneity of DNA cell cycle variables in breast cancer measured by flow cytometry. J Clin Pathol 1996; 49: 931–937.
Braun S, Hepp F, Sommer HL, Pantel K . Tumor antigen heterogeneity of disseminated breast cancer cells: implications for immunotherapy of minimal residual disease. Int J Cancer 1999; 84: 1–5.
Rummukainen JK, Salminen T, Lundin J, Joensuu H, Isola JJ . Amplification of c-myc oncogene by chromogenic and fluorescence in situ hybridization in archival breast cancer tissue array samples. Lab Invest 2001; 81: 1545–1551.
Simon R, Nocito A, Hubscher T, Bucher C, Torhorst J, Schraml P, et al. Patterns of HER-2/neu amplification and overexpression in primary and metastatic breast cancer. J Natl Cancer Inst 2001; 93: 1141–1146.
Acknowledgements
This work was funded by a grant from the National Health Group of Singapore, NHG-RPR/01121. DHZ and ED are funded by a grant from The Health Service Development Program, Ministry of Health, Singapore. The skillful technical assistance of Mr. Ow Cheok Kee and Mr. Tan Tee Chok is greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, D., Salto-Tellez, M., Putti, T. et al. Reliability of Tissue Microarrays in Detecting Protein Expression and Gene Amplification in Breast Cancer. Mod Pathol 16, 79–85 (2003). https://doi.org/10.1097/01.MP.0000047307.96344.93
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.MP.0000047307.96344.93
Keywords
This article is cited by
-
Statin use, candidate mevalonate pathway biomarkers, and colon cancer survival in a population-based cohort study
British Journal of Cancer (2017)
-
Ki67 expression in invasive breast cancer: the use of tissue microarrays compared with whole tissue sections
Breast Cancer Research and Treatment (2017)
-
Evaluation of PTGS2 Expression, PIK3CA Mutation, Aspirin Use and Colon Cancer Survival in a Population-Based Cohort Study
Clinical and Translational Gastroenterology (2017)
-
Impact of tissue sampling on accuracy of Ki67 immunohistochemistry evaluation in breast cancer
Diagnostic Pathology (2016)
-
Reply to: Comment on ‘Tumour antigen expression in hepatocellular carcinoma in a low-endemic western area’
British Journal of Cancer (2016)
