
Abstract
We describe two cases of peripheral primitive neuroectodermal tumor–Ewing's sarcoma (PNET-ES) arising intracranially in the leptomeninges. Both tumors exhibited a primitive undifferentiated round-cell morphology. Immunohistochemical stains revealed strong membrane expression of CD99 in both cases. A t(11;22)(q24;q12) could be demonstrated with reverse transcriptase–polymerase chain reaction in one case, whereas fluorescence in situ hybridization analysis performed in the second case showed a rearrangement of the EWS gene. The occurrence of PNET-ES at this site is very unusual. Immunophenotypical as well as genetic analysis play a key role in the diagnosis and the distinction from central PNET.
Similar content being viewed by others

INTRODUCTION
Extracerebellar primitive neuroectodermal tumors (PNETs) are uncommon central nervous system (CNS) tumors, affecting primarily children and young adults. They are mostly intraparenchymal, located supratentorially or, less frequently, in the spinal cord, but primary localization of these tumors in the meninges has been reported (1).
Histologically, these tumors are composed of small undifferentiated neuroectodermal cells and frequently show immunohistochemical and/or electron-microscopic features of divergent neuronal or glial differentiation (2). In contrast with posterior fossa PNET or medulloblastoma, occurrence of isochromosome 17q is very rare (2). A limited number of miscellaneous nonrandom cytogenetic gains and losses has been reported in the few cases that have been successfully karyotyped (3, 4).
PNETs arising outside the CNS, most frequently in the deep soft tissues of the trunk and lower limbs, are now considered part of a spectrum of round cell sarcoma, including Ewing's sarcoma (ES) and peripheral PNET (5). These tumors typically express high amounts of the MIC2 antigen (CD99) (6) and exhibit highly characteristic chromosomal translocation that results in the fusion of the EWS gene with any of several members of the ETS family of transcription factors, leading to oncogenic activation of the EWS gene (7).
We herein describe two cases of primary meningeal PNET-ES. Both tumors exhibited morphologic, immunophenotypic, and molecular genetic features diagnostic of peripheral PNET-ES, a tumor distinct from the relatively more common central PNET.
MATERIAL AND METHODS
Case Material
We studied two patients, a 17-year-old man and a 12-year-old boy. The clinical and imaging features of the cases are summarized in Table 1. In both cases, because of the imaging features of the lesion, dura based, with iso/hypointense T1 signal and intense contrast enhancement, the preoperative diagnosis of meningioma was suggested (Figs. 1 and 2). After the diagnosis of PNET-ES, both patients received adjuvant therapy. The first patient received local radiotherapy (30 × 2 Gy) after his first surgery and systemic chemotherapy [Cysplatinum (30 mg/m2/d), Iphosphamide (2000 mg/m2/d) and Etoposide (100 mg/m2/d)] followed by craniospinal radiation (22 × 1.6 Gy + 10 × 2 Gy boost on the left CPA) after the second surgery. He has no signs of progression at 12 months after the last surgery. The second patient was treated according to the Italian Pediatric Oncology Group protocol for CNS PNET/medulloblastoma including two preRT chemotherapy courses consisting of systemic and intrathecal administration of Methotrexate (8 g/m2 on Day 1), followed by four courses of Carboplatin (800 mg/m2) on Day 8 and VP16 (150 mg/m2) on Days 8, 9, and 10, the course to be repeated every 28 days. Subsequently, the patient was treated with craniospinal RT (36 Gy + 18 Gy boost on the primary site), followed by four courses of Cisplatin (70 mg/m2), Lomustine (80 mg/m2), and Vincristine (1.5 mg/m2; 8). This patient is alive and well without evidence of disease, 27 months after the first diagnosis.

MRI (T1 weighted, after Gadolinium injection): parasagittal, polylobulated mass with broad base on the dura; intense contrast enhancement is present (Case 1).

MRI (T1 weighted, after Gadolinum injection): a right parietal, parasagittal mass, strongly and diffusely enhancing after Gadolinum injection (Case 2). Dural “tails” are present, suggesting the preoperative diagnosis of meningioma.
Pathological Studies
Surgical specimens of all tumors were fixed in 10% formaldehyde, embedded in paraffin, sectioned and stained with hematoxlin and eosin, reticulin stain, and periodic acid–Schiff, with and without diastase digestion. An extensive panel of immunohistochemical stains was performed, including the following antibodies: vimentin (monoclonal [MC]; Amersham; 1/20), keratin (MC; Immunotech; 1/50), CD45 (MC; DAKO; 1/50), glial fibrillary acidic protein (GFAP; polyclonal; DAKO; 1/300), synaptophysin (MC; DAKO; 1/10), neurofilament (MC; Monosan; 1/10), S100 protein (PC; DAKO; 1/300), epithelial membrane antigen (MC; DAKO; 1/100), desmin (MC; DAKO; 1/100), and CD99 (MIC2, MC; DAKO, 1/200; 013 MC; Signet, 1/100). Negative and positive controls were employed throughout. Formaldehyde-fixed tissue from left PCA tumor of Case 1 was embedded in Epon for ultrastructural investigation.
Molecular Studies
In Case 1, RNA was extracted from frozen tissue available from the left CPA tumor. Reverse transcription polymerase chain reaction (RT-PCR) was performed, using previously described oligonucleotide primers for Exon 7 of EWS (5′-TCCTACAGCC- AAGCTCCAAGTC-3′) as the forward primer and Exon 9 of FLI1 (5′-ACTCCCCGTTGGTCCCCTCC-3′) as the reverse primer (9). In Case 2, fluorescence in situ hybridization (FISH) was performed using commercially prepared reagents (Oncor Tissue Kit, Oncor, Gaithersburg, MD) according to the manufacturer's recommendations. Four-micrometer-thick paraffin-embedded sections were prepared on silane-coated slides. After deparaffinization, tissue sections were placed in 30% pretreatment solution, digested in proteinase K, dehydrated, and denatured. Dual-color FISH was performed using DNA probes consisting of yeast artificial chromosome contigs that were mapped immediately centromeric (labeled with digoxin and amplified with FITC anti-digoxigenin) and telomeric (labeled with biotin and detected with avidin-rhodamine) to the EWS gene on chromosome 22 (10). Slides were counterstained with DAPI/Antifade (0.5 mg/mL; Oncor) and evaluated using a Zeiss Axioscope fluorescence microscope (Carl Zeiss, Jena, Germany). Tumor cell nuclei that showed a split of at least half a nuclear diameter for a centromeric–telomeric EWS region FISH signal pair were scored as positive for a rearrangement in chromosome 22q12. Hybridization signals were scored for 50 nuclei.
RESULTS
Pathology
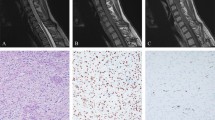
Grossly, both frontal tumors showed a broad implantation base on the dura. They were well circumscribed, somewhat lobulated, and measured 4.5 and 5 cm in greatest dimension. On cut surface, areas of hemorrhage and/or necrosis were present. Recurrent tumor from Patient 1 was removed in a piecemeal fashion. Microscopically, sheets and compact nests of uniform small blue cells with scant cytoplasm were seen in all lesions (Fig. 3A and 3B). Well-formed rosettes were absent. The nuclei were round to oval with finely dispersed chromatin and a small nucleolus. Mitotic figures were numerous. All tumors were highly vascularized and showed a dense pericellular reticulin network. Most of the tumor cells contained periodic acid-Schiff–positive, diastase-sensitive material consistent with glycogen.

Case 2: low power microscopic view, showing sheets of monotonous small round cells (A). High power microscopic view, highlighting an area of necrosis and several mitotic figures (B). The tumor cells display a diffuse, strong, membrane expression of CD99 (C). FISH with EWS centromere/EWS telomere probe set of Case 2. Split signals indicate EWS region rearrangement (D).
Immunohistochemically, both tumors demonstrated diffuse, strong positivity for CD99 (Fig. 3c). Focal positivity for S100 protein was observed in Case 1, whereas focal synaptophysin as well as neurofilament expression was detected in Case 2. Expression of keratin, CD45, GFAP, desmin, and EMA was not detected in either tumor specimen.
Electron-microscopic studies performed on the left CPA tumor from Case 1 confirmed the limited differentiation of the tumor cells, with round irregular nuclei, finely distributed chromatin, and a small nucleolus. The cytoplasm contained scant organelles and large amounts of glycogen. No dense core granules, myofilaments, or basal membranes were seen. Therewere a few primitive junctional complexes.
Molecular Studies
FISH analysis in Case 2 revealed rearrangement of the EWS region on chromosome 22q12 in 17 of 50 cells (Fig. 3d). This result is consistent with a translocation involving the chromosome 22q12 region, characteristic of PNET-ES.
RT-PCR in Case 1 revealed the EWS/FLI1 fusion transcripts of the t(11, 22)(q24;q12) translocation (Fig. 4).

Result of the RT-PCR for the detection of the EWS/FLI1 fusion transcript in Case 1. Lane M: molecular marker; Lane 1: positive control; Lane 2: patient's sample; Lane 3: negative control.
DISCUSSION
Central PNET and peripheral PNET-ES exhibit characteristic immunophenotypical as well as genetic features which allow their distinction from other small round cell tumors. We have herein documented two cases of intracranial, dura-based tumors showing morphologic as well as molecular genetic features of peripheral PNET-ES. Localization of primitive neuroectodermal tumors to the meninges is exceptional (11, 12, 13, 14, 15, 16). Among previously reported cases, two patterns of meningeal involvement are described. The first is characterized by a diffuse involvement of the cranial and spinal leptomeninges in the absence of a primary intraparenchymal or meningeal tumor (11, 12, 13). No information is available regarding the CD99 expression and the t(11, 22) status of these tumors. The second is characterized by a localized dural-based mass, mimicking meningioma, similar to our cases. Only three such cases with features of PNET-ES (14, 15, 16) have been reported, two of which with a proven t(11, 22) (15, 16) (Table 2). An additional case in a 2-month-old girl most likely represents extension to the meninges of a PNET-ES arising in the skull (17). A case of extracerebral neuroblastoma arising from the convexity dura mater has also been reported (18), which might represent another example of this entity. However, without immunohistochemical and molecular studies, this cannot be established. In our cases, the diagnosis has been proved histologically, ultrastructurally (in Case 1), immunohistochemically, and at the molecular level.
The MIC2 gene product (CD99) is highly expressed immunohistochemically in nearly all peripheral PNET-ES (19), a feature that, although highly sensitive, is not specific for PNET-ES. CD99 immunopositivity can also be detected also in other small, blue round cell tumors (19, 20) in which, however, the pattern of staining is often cytoplasmic, rather than the distinct membranous staining typical of PNET-ES. Central PNETs are reported to be negative for CD99 staining (21, 22).
The chromosomal translocation t(11, 22)(q24;q12) is found in >90% of peripheral PNET-ES and appears to be characteristic (7, 23, 24). It results in the fusion of the EWS gene with a truncated transcription factor FLI1 on 11q24, causing oncogenic conversion of the EWS gene. The t(11, 22) translocation is not found in primary cerebral and cerebellar PNET (25, 26).
The cases reported in this paper, as well as the previously reported cases, appear to represent genuine examples of occurrence of PNET-ES of the dura. Although PNET-ES has a predilection for bone and soft tissue, it can arise virtually at any location. The distinction between peripheral PNET-ES and central PNET may be clinically important. The long clinical course observed in three of five cases with available follow-up parallels the long-term disease-free survival reported in up to 45 to 60% of PNET-ES cases (27). Among patients with intracranial central PNETs, long-term survival is uncommon (28). Location and circumscription of the tumors, which allowed gross total resection, may have played an important role in the outcome. Although at present the knowledge of the genetic background of the tumor may have not direct bearing on treatment and/or outcome, distinction of these lesions on genetic analysis may become important for future treatment protocols given the recognized sensitivity of PNET-ES to chemotherapy.
References
Hart MN, Earle KM . Primitive neuroectodermal tumors of the brain in children. Cancer 1973; 32: 890–897.
Rorke LB, Hart MN, McLendon RE . Supratentorial primitive neuroectodermal tumor (PNET).In: Kleihues PCW, ed. Tumors of the nervous system. Lyon, France: IARC Press; 2000.p. 141–144.
Burnett ME, White EC, Sih S, von Haken MS, Cogen PH . Chromosome arm 17p deletion analysis reveals molecular genetic heterogeneity in supratentorial and infratentorial primitive neuroectodermal tumors of the central nervous system. Cancer Genet Cytogenet 1997; 97: 25–31.
Russo C, Pellarin M, Tingby O, Bollen AW, Lamborn KR, Mohapatra G, et al. Comparative genomic hybridization in patients with supratentorial and infratentorial neuroectodermal tumors. Cancer 1999; 86: 331–339.
Dehner LP . Peripheral and central primitive neuroectodermal tumors: a nosologic concept seeking a consensus. Arch Pathol Lab Med 1986; 110: 997–1005.
Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M . MIC2 is a specific marker for Ewing's sarcoma and peripheral primitive neuroectodermal tumors. Cancer 1991; 67: 1886–1893.
Whang-Peng J, Triche TJ, Knutsen T, Miser J, Douglass EC, Israel MA . Chromosome translocation in peripheral neuroepithelioma. N Engl J Med 1984; 311: 584–585.
Pezzotta S, di Montezemolo C, Knerich R, Arrigoni M, Perilongo G, Madon E . CNS-85 trial: a cooperative pediatric CNS tumour study—results of treatment of medulloblastoma patients. Childs Nerv Syst 1996; 12: 87–96.
Barr FG, Chatten J, D'Cruz CM, Wilson AE, Nauta LE, Nycum LM, et al. Molecular assays for chromosomal translocations the diagnosis of pediatric soft tissue sarcoma's. JAMA 1995; 273: 553–557.
Davison JM, Morgan TW, Hsi BL, Xiao S, Fletcher JA . Subtracted, unique-sequence, in situ hybridization: experimental and diagnostic applications. Am J Pathol 1998; 153: 1401–1409.
Fujii M, Orita T, Nishizaki T, Aoki H, Tanaka D . Primitive neuroectodermal tumor of the leptomeninges. Neuroradiology 1991; 33: 260–263.
Mendel RC, Pollay M, Bobele GB, Leech RW, Brumback R . Primary primitive neuroectodermal tumor of the leptomeninges. J Child Neurol 1996; 11: 404–407.
Szpak GM, Papierz W, Liberski PP, Kulczycki J, Kryst-Widzgowska T, Dymecki J . Primitive neuroectodermal tumor (PNET). A case report. Folia Neuropathol 1995; 33: 35–40.
Katayama Y, Kimura S, Watanabe T, Yoshino A, Koshinaga M . Peripheral-type primitive neuroectodermal tumor arising in the tentorium. Case report. J Neurosurg 1999; 90: 141–144.
Papotti M, Abbona G, Pagani A, Monga G, Bussolati G . Primitive neuroectodermal tumor of the meninges: an histologic, immunohistochemical, ultrastructural and cytogenetic study. Endocr Pathol 1998; 3: 275–280.
Antunes NL, Lellouch-Tubiana A, Kalifa C, Pierre-Kahn A, Rosenblum MK . Intracranial Ewing sarcoma/‘peripheral’ primitive neuroectodermal tumor of dural origin with molecular genetic confirmation. J Neurooncol 2001; 51: 51–56.
Yoshizato K, Yoshioka S, Tamai T, Tsuji K, Nishio S . Intracranial primitive neuroectodermal tumor in an infant: a case report. No Shinkei Geka 1999; 27: 243–248.
Ishii N, Sawamura Y, Aoki T, Nagashima K, Kashiwaba T, Abe H . Intracranial extracerebral neuroblastoma originating from the convexity dura mater. Neurol Med Chir (Tokyo) 1996; 36: 19–22.
Hasegawa SL, Davison JM, Rutten A, Fletcher JA, Fletcher CDM . Primary cutaneous Ewing's sarcoma. Immunophenotypic and molecular cytogenetic evaluation of five cases. Am J Surg Pathol 1998; 22: 310–318.
Dei Tos AP, Wadden C, Calonje E, Sciot R, Pauwels P, Knight JC, et al. Immunohistochemical demonstration of p30/32 MIC2 (CD99) in synovial sarcoma. A potential cause of diagnostic confusion. Appl Immunohistochem 1995; 3: 168–173.
Gyure KA, Prayson RA, Estes ML . Extracerebellar primitive neuroectodermal tumors: a clinicopathologic study with bcl-2 and CD99 immunohistochemistry. Ann Diagn Pathol 1999; 3: 276–280.
Batsakis JG, El-Naggar AK . Ewing's sarcoma and primitive neuroecodermal tumors: cytogenetic cynosures seeking a common histogenesis. Adv Anat Pathol 1997; 4: 207–220.
Aurias A, Rimbaut C, Buffe D, Dubousset J, Mazabraud A . Chromosomal translocations in Ewing's sarcoma. N Engl J Med 1983; 309: 496–498.
Turc-Carel C, Philip I, Berger MP, Philip T, Lenoir GM . Chromosomal translocations in Ewing's sarcoma. N Engl J Med 1983; 390: 497–498.
Jay V, Peinkowska M, Becker L, Zielenska M . Primitive neuroectodermal tumors of the cerebrum and cerebellum: absence of t(11;22) translocation by RT-PCR analysis. Mod Pathol 1995; 8: 488–491.
Jay V, Zielenska M, Lorenzana A, Drake J . An unusual cerebellar primitive neuroectodermal tumor with t(11;22) translocation: pathological and molecular analysis. Pediatr Pathol Lab Med 1996; 16: 119–128.
Schmidt D, Hermann C, Jurgens H, Harms D . Malignant peripheral neuroectodermal tumor and its necessary distinction from Ewing's sarcoma. A report from Kiel pediatric tumor registry. Cancer 1991; 68: 2251–2259.
Dirks PB, Harris L, Hoffman HJ, Humphreys RP, Drake JM, Rutka JT . Supratentorial primitive neuroectodermal tumors in children. J Neurooncol 1996; 29: 75–84.
Acknowledgements
The authors wish to thank Dr. Jonathan Fletcher, Department of Pathology, Brigham & Womens Hospital, Boston, Massachusetts, for kindly providing EWS probes for FISH analysis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dedeurwaerdere, F., Giannini, C., Sciot, R. et al. Primary Peripheral PNET/Ewing's Sarcoma of the Dura: a Clinicopathologic Entity Distinct from Central PNET. Mod Pathol 15, 673–678 (2002). https://doi.org/10.1038/modpathol.3880585
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880585
Keywords
This article is cited by
-
Primary, Dural-based, Ewing sarcoma in a pediatric patient: presentation of a rare tumor entity with literature review
Child's Nervous System (2023)
-
Primary intracranial Ewing sarcoma/ peripheral primitive neuroectodermal tumor, an entity of unacquaintance: a series of 8 cases
Child's Nervous System (2021)
-
Ewing sarcoma and Ewing-like tumors
Virchows Archiv (2020)
-
Primary intracranial extraosseous Ewing’s sarcoma
Child's Nervous System (2019)
-
Dura-based mass in a young boy
Child's Nervous System (2017)
