
Abstract
PTLD is a well-recognized complication of organ transplantation. Large series of heart, renal, and liver transplants have been examined for the incidence and behavior of PTLD. However, reports of the incidence and characteristics of PTLDs in lung transplant (LTx) patients are few. We report our experience with PTLDs in a large series of LTx recipients at a single institution and compare them to other solid organ transplant recipient PTLDs seen at our institution. Twenty-eight patients were found to have PTLD, of whom 8 were lung transplant recipients. We evaluated nine PTLD specimens from these 8 patients for their histology, immunophenotype (CD20, CD3, EBV-LMP1), EBER status by in situ hybridization, and clinical features. The incidence of PTLD was 3.3% (8/244 patients). The time to development of PTLD, after transplant, was short (median time, 7 mo). All were of B-cell lineage. Overall, EBV was demonstrated in 77.7% (7 of 9 specimens) of PTLDs. All specimens tested for clonality were found to be monoclonal. Five patients died, with a median time to death of only 4.6 months. PTLDs in LTx patients are EBV-associated B-cell, predominantly monoclonal lymphoid lesions similar to other solid organ transplant PTLDs. Compared with other solid organ transplant recipients with PTLD at our institution, PTLDs in LTx patients have a propensity to involve the transplanted organ (P = .001, Fisher's exact test), occur earlier after transplant (P = .003, Wilcoxon test), and have a shorter survival (P = .002, log rank test). Reasons for this may include the relatively higher level of immunosuppression required in these patients and limited options in decreasing it. Although the incidence is low, careful early monitoring of lung transplantation patients is warranted because of the poor prognosis of patients developing this complication.
Similar content being viewed by others

INTRODUCTION
Posttransplant lymphoproliferative disorder (PTLD) is a well-known complication in solid organ transplant patients (1, 2, 3, 4). The incidence of PTLDs in transplant patients ranges from <2 to 10% (5, 6). These are a heterogeneous group of disorders that have varying morphology, clinical manifestations, biological behavior, and response to treatment. PTLDs are commonly associated with Epstein-Barr virus (EBV) infection, implicating it in the pathogenesis of these lesions (2, 7, 8, 9, 10). Known risk factors include the patient's age, transplanted organ, EBV status before transplant, and immunosuppressive drugs (11, 12).
Over the past decade, numerous attempts have been made to classify these lesions using morphology, immunohistochemistry, and monoclonality, but none of these tools alone or in combination accurately predicts biological behavior (13, 14). Monoclonality is considered an adverse prognostic feature; however, some monoclonal PTLDs regress with reduction in immunosuppression. Conversely, a subset of polyclonal lesions has a more aggressive course (14). More recently other molecular genetic studies have detected oncogene alterations and BCL-6 mutations that have been shown to be useful prognostic indicators (15, 16).
The incidence of PTLDs varies depending on the transplanted organ (11).The characteristics of PTLDs have been studied in large series of heart, kidney, and liver transplant patients (3, 12, 17, 18, 19, 20, 21). Large series reporting the incidence and presentation of PTLDs exclusively in lung transplant (LTx) patients are few (12, 22, 23, 24). We recently encountered two PTLDs in LTx patients that occurred early after transplantation and demonstrated an aggressive clinical behavior. On review of the literature, we found a varied experience, with some reporting a high incidence of PTLD in LTx patients, whereas others found a much lower incidence (12, 22, 23, 24). We reviewed our experience with PTLDs in LTx patients at the Cleveland Clinic to determine whether these PTLDs have distinctive clinical or pathologic features compared with those in other solid organ recipients.
MATERIALS AND METHODS
The files from the department of Anatomic Pathology at the Cleveland Clinic Foundations were searched for cases of PTLD occurring in solid organ transplant patients. All cases of PTLDs regardless of the organ transplanted were retrieved for evaluation. Four thousand thirty-three patients underwent solid organ transplantation (244 lung, 564 liver, 2332 kidney, and 893 heart) at the Cleveland Clinic at the time of our search. Twenty-eight patients were found to have PTLD, of whom 8 were recipients of LTxs. Nine specimens from the eight LTx patients consisted of seven paraffin-embedded tissue biopsy blocks and two cytology specimens. One patient had a recurrence, accounting for the one additional specimen. The remaining 20 patients were from other solid organ transplant procedures: kidney (n = 8), heart (n = 7), and liver (n = 5). Paraffin-embedded tissue blocks were available for 21 specimens (1 recurrence) from these patients. Although we are certain that all PTLDs were captured from the LTx patients, the PTLDs from the other solid organ transplant patients may not be complete because of limitations in searching before 1988. Clinical features including initial disease for transplant, time to PTLD, site of PTLD, stage of the patient, recurrence, immunosuppression regimen, and type and response to treatment, were evaluated for all PTLDs. One LTx patient (Patient 3) was previously reported (25). Autopsies were performed in five cases (1 LTx patient and 4 non-LTx patients).
Statistics
Comparative analysis of clinical, morphological, and immunohistochemical parameters between LTx and other solid organ transplants was done using either the chi-square test or Fisher's exact test, for categorical variables, or the Wilcoxon test, for continuous variables. Overall survival was estimated using the Kaplan-Meier method and compared between LTx and other solid organ transplants via log-rank test. P < .05 was used to indicate statistical significance.
Immunophenotyping
Immunohistochemistry (IHC) using a standard streptavidin-biotin complex technique was performed on paraffin-embedded sections and cell blocks using an automated stainer (Ventana Medical Systems, Tucson, AZ). Lineage was established in all cases either by CD3 and CD20 immunostaining or by flow cytometry. All cases were stained for LMP-1. Kappa and lambda light chain analysis was performed in a subset of non-LTx PTLDs either by IHC or flow cytometry at the time of initial diagnosis. Only Patient 8 among the LTx PTLD was tested for immunoglobulin light chain restriction (by IHC), and monoclonality was established by molecular techniques in the other cases. The cytology specimens were not tested for clonality because of insufficient tissue. Additionally CD30, CD43, and CD79a were also performed in some LTx PTLDs at the time of diagnosis, and pertinent results are reported in Table 1. The identity and source of antibodies is summarized in Table 2.
Four-color flow-cytometric immunophenotyping was performed using a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA). Monoclonal antibodies to CD3-allophycocyanin (APC), CD4-fluorescein isothiocyanate (FITC), CD5 peridinin chlorophyll protein (PerCP), CD8 phycoerythrin (PE), CD10 FITC, CD19 APC, CD20 PE, CD45 PerCP, CD79a PE, kappa FITC, and lambda PE were used to stain 2 × 10−6 cells. Ten thousand cells were acquired per tube using CellQuest™ software (Becton Dickinson), and the results were analyzed using Paint-a-gate™ (Becton Dickinson).
Molecular Genetics
Molecular genetic analysis for the detection of immunoglobulin heavy chain gene rearrangement was done on seven specimens from six patients, using either Southern blot analysis (SBA; four specimens) or polymerase chain reaction (PCR; three specimens) techniques.
For the SBA, DNA was isolated from the frozen tissue by incubation with DNAZOL (Molecular Research Center, Cincinnati, OH) according to the manufacturer's protocol. The DNA was quantified using spectrophotometric measurement and digested with restriction endonucleases. BamHI/HindIII and EcoRI (Gibco BRL, Grand Island, NY) restriction endonucleases were used. The DNA fragments were separated using 1% agarose gel (FMC Corp. Philadelphia, PA) electrophoresis. The gel was stained with ethidium bromide (Sigma Diagnostics, St. Louis, MO) and viewed under ultraviolet light. The separated DNA fragments were denatured, transferred to a nylon membrane, hybridized to fluorescent-labeled probe (kappa −2.5Kb and JH −2.5Kb; both DAKO), and detected by chemiluminescence using standard manufacturer's (DAKO) protocol.
PCR for VDJ gene rearrangements was performed according to methods described elsewhere (26). Briefly DNA was extracted from paraffin-embedded tissue by initial dewaxing using Autodewaxer (Research Genetics, Inc., Huntsville, AL) and tissue digestion using 200 g/mL of Proteinase K (Boehringer Mannheim, Indianapolis, IN) and Tris buffer at 55° C overnight. VDJ PCR framework II and III primer sets were used. The primer sequences are summarized in Table 2. Bβeta-globin primer set was used to assess whether amplifiable DNA was present. All tests were run with positive and negative controls. The product was and analyzed on a 6% polyacrylamide gel (Zaxis, Hudson, OH), stained with ethidium bromide (sigma), and viewed by ultraviolet transilluminator.
Chromogenic In Situ Hybridization
EBV-encoded RNA (EBER) by in situ hybridization (ISH) was using an automated stainer (Ventana). Unstained paraffin sections on electrostatically charged slides were baked overnight at 60° C, deparaffinized in two changes of xylene for 5 minutes each, followed by absolute ethanol (two 10-min changes each). Slides were then washed in distilled water for 5 minutes. Sections fixed in B5 or Hollande's were treated with Lugol's iodine (Fisher, Pittsburgh, PA) and sodium thiosulphate (Allegiance, McGaw Park, IL) to remove heavy metal deposits. The sections were then enzyme digested in 100 μL of Protease I (Ventana Medical Systems, Tucson, AZ) at 37° C for 8 minutes and dehydrated in graded alcohols. The reaction was stopped in distilled water for 5 minutes. Ten microliters of biotin-labeled oligonucleotide (EBER) probe (Table 3), in hybridization solution (Sigma) consisting of 50% deionized formamide, 20 × standard saline citrate (SSC), and dextran sulfate was applied to the section and hybridized overnight at 37° C in a humidified chamber. Stringency wash of 2 × SSC was performed for 5 minutes at room temperature. Hybridization detection was performed using a streptavidin-alkaline phosphatase detection according to manufacturer's (Ventana) instructions. This consisted of sequential application of 100 μL of mouse antibiotin antibody and enhanced alkaline phosphatase-labeled streptavidin, followed by color development with a 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium system (Ventana). Slides were counterstained with Nuclear Fast Red (Newcomer Supply, Middletown, WI) for 8 minutes, dehydrated, and mounted with Cytoseal 60 (Stephens Scientific).
A few cases in the non-lung Tx cohort that were negative on initial EBER testing but had positive LMP-1 staining were retested with INFORM® EBER probe (Ventana). The staining was done on the Discovery automated ISH instrument (Ventana) per manufacturer protocol.
High-Throughput Tissue Microarray
A high throughput tissue microarray (TMA; Beecher, Silver Spring, MD) was constructed from specimens with suitable tissue blocks. The microscopic slides were reviewed, and the sections with tumor were microarrayed using a 1.5-mm-diameter stylet. Two such blocks were constructed with appropriate controls incorporated in them and were used for immunohistochemical analysis. For specimens with small amounts of tissue unsuitable for TMA, and in cases in which staining was uncertain, routine unstained slides were used for immunohistochemical analysis.
RESULTS
Clinical Findings
Eight of 244 LTx patients developed PTLDs (3.3%). The mean age was 51 years (range, 31–62 y). Four patients were male, and four were female. The median time to onset of PTLD was 7 months. The initial site of PTLD included the transplanted lung (n = 5), colon (n = 1), small bowel (n = 1), and skin (n = 1). Most of the LTx patients presented with low-stage disease (62.5% were Stage IE). Clinically, none of the patients had primary lymph node involvement by PTLD. Patient 8 presented with multiple synchronous tumors in the colon. The clinical data for LTx patients is summarized in Table 1.
We identified 20 non-LTx PTLD patients. The mean age of presentation was 49 years (heart, 52 y; kidney, 47 y; liver, 46 y). The median time to development of PTLD after transplantation was 40.5 months, and low stage was also common (43% were Stage IE, and 14% were Stage IIE). The most common site of primary involvement by PTLD was lymph node. In contrast to LTx recipients, none of the patients developed PTLD in the allograft organ (P = .001, Fisher's exact test). The clinical data for non-LTx patients is summarized in Table 4.
There appeared to be no significant difference in age (P = .99, Wilcoxon test) or stage between lung and non-LTx patients (P = .44, chi-square test). However, the median time to onset of PTLD after LTx was significantly shorter in the LTx patients compared with in other solid organ recipients. (P = .003, Wilcoxon test).
The immunosuppression protocols for the lung and other solid organ transplant patients are summarized in Table 5. In general, LTx recipients received a higher degree of immunosuppression, in particular in the forms of cyclosporine and methylprednisolone, compared with other solid organ transplant recipients. Lung transplant patients do not receive anti-lymphocyte antibody therapy as part of their immunosuppression protocol.
Morphologic Features
All cases were classified according to the WHO classification (27). Of the LTx PTLDs, three cases were polymorphic (Fig. 1), and five were monomorphic PTLDs (Fig. 2; Table 1). The monomorphic PTLDs were characterized by monotonous sheets of atypical lymphoid cells similar to a diffuse large B-cell lymphoma. A spectrum of B-cell maturation including small and large lymphocytes, plasma cells, and atypical immunoblasts was characteristic of the polymorphic PTLDs. Patient 8 (considered monomorphic in Table 1) actually had multiple synchronous colon tumors demonstrating monomorphic as well as polymorphic and plasmacytoma-like PTLDs. The plasmacytoma-like PTLD was composed of sheets of both mature and atypical plasma cells with variably prominent nucleoli (Fig. 3). Patient 2 initially presented with a polymorphic PTLD and subsequently had a recurrence that was a monomorphic PTLD. The two FNA specimens had cell blocks that contained a few tissue fragments composed of large atypical lymphoid cells with prominent nucleoli that were best classified as monomorphic PTLD.

An example of a polymorphic PTLD showing a spectrum of B-cell maturation including centrocyte and centroblast-like cells and plasma cells.

A monomorphic PTLD demonstrating sheets of pleomorphic, atypical lymphoid cells.

A plasmacytoma-like PTLD in the colon consisting of a diffuse infiltrate of both mature and occasional atypical plasma cells.
Of the 21 specimens from the 20 non-LTx PTLD patients, 15 were monomorphic, 2 were polymorphic, 2 were plasmacytoma-like, and 1 was a Hodgkin lymphoma-like PTLD (Table 4). The Hodgkin lymphoma-like PTLD contained RS-like cells in the background of a diffuse inflammatory infiltrate composed of eosinophils, neutrophils, small lymphocytes, and occasional plasma cells similar to the mixed cellularity type of Hodgkin lymphoma. One patient who presented with a polymorphic tumor had a recurrence that was monomorphic. There was no statistically significant difference in the incidence of morphologic subtypes in lung and non-LTx patients (P = .28 by chi-square test).
Immunophenotypic Features
All cases in LTx patients were B-cell lineage and expressed CD20 (Fig. 4). Two cases showed coexpression of CD43, five had CD79a expression, and one case was CD30 positive (Table 1). Patient 8, who had multiple colonic PTLDs, showed lambda light chain restriction at the time of diagnosis. Further testing showed that other colonic tumors had kappa light chain restriction.

A monomorphic PTLD showing diffuse CD 20 positivity.
Among the non-LTx PTLDs 17 of 21 specimens were assessed for light chain restriction (4 by IHC, 10 by flow cytometry, and 3 by both methods). Of these, 3 were kappa monotypic, 3 were lambda monotypic, 2 were polytypic, and 7 were surface Ig light chain negative; in 2 specimens, the stain was indeterminate. One of the non-LTx PTLDs was a T-PTLD in a patient who had a kidney transplant, and Ig light chain restriction analysis was not relevant in this case. Ig light chain restriction was also not tested for in the Hodgkin-disease like PTLD. All other non-LTx PTLDs were of B-cell lineage.
Molecular Genetics
Among LTx patient PTLDs clonality was evaluated by SBA or PCR in seven specimens, all of which were found to be monoclonal (Table 1). Neither test for clonality was performed on the FNA specimens (n = 2). Of the 21 tumors from the 20 non-LTx patients, 12 were monoclonal and seven were polyclonal, whereas two cases had insufficient tissue for analysis (Table 4).
EBV Analysis
EBER ISH was performed on paraffin-embedded sections of 7 specimens from 6 LTx patients and was positive in all cases (Table 1, Fig. 5). EBV LMP-1 was tested on all specimens and was found to be positive in 6 of 9 specimens (Table 1). Five specimens were positive for both EBER and LMP-1. Two were positive for EBER but negative for LMP-1.

Dark blue nuclear staining pattern of EBER using chromogenic in situ hybridization.
Fifteen of the 21 specimens (71%) among non-LTx patients were positive for EBER. Fourteen of the EBER positive specimens also expressed LMP-1. For one EBER-positive specimen, the LMP-1 was noninterpretable (Table 4). One EBER-negative specimen was LMP-1 positive, possibly because of either a false-positive LMP-1 because of cross-reactivity (28) or inability of the EBER probe to detect the RNA. There was no significant difference in EBV positivity by ISH when LTx PTLDs were compared with those in other solid organ recipients (P = .13 by chi-square test).
Outcome
Treatment of the LTx patients was not uniform and consisted of varying combinations of reduction of immunosuppression, radiotherapy, and chemotherapy. Details of therapy are listed in Table 3. Follow-up time for the LTx patients ranged from 0.4 to 20 months (mean, 5.2 mo). Five of the 8 patients (62.5%) died of complications of the disease. The median time to death was 4.6 months. Two patients were alive with evidence of disease. One patient was lost to follow-up (Table 1).
Follow-up time for non-LTx patients ranged from 1–58.1 months (mean 21.7 mo). At last follow-up, four patients had died of complications of the disease (20%), two patients had died of unrelated causes (10%), five were alive with evidence of disease (25%), and nine were alive with no evidence of disease (45%; Table 4).
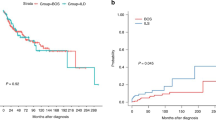
The overall survival after PTLD of LTx patients was compared with that of other solid organ transplant patients by Kaplan Meier survival analysis. LTx patients had a much shorter survival when compared with the rest of the cohort (P = .002, log-rank test; Fig. 6).

Kaplan-Meier survival plot comparing survival between lung and other solid organ transplant recipients.
Autopsy Results
Autopsies were performed on two LTx-PTLD patients. Autopsy on Patient 2 showed residual PTLD in both the right lung and colon. Multifocal nodules of PTLD were also found in the leptomeninges, cerebrum and cerebellum. Patient 3, who initially presented with cutaneous PTLD was found to have PTLD in the left kidney. All other organs were uninvolved. Additionally biopsies of the cutaneous lesions on her lower limb demonstrated atypical lymphoid cells consistent with residual PTLD.
All three non-LTx patients who had autopsies were heart transplant recipients. Two patients who died of complications of the disease while undergoing treatment had no evidence of PTLD at autopsy. The third patient had residual disease at the initial site of presentation.
DISCUSSION
PTLDs are a well-recognized complication of organ transplantation and are generally EBV-associated B-cell proliferations. The incidence of PTLDs appears to depend on several factors, including age, pretransplantation EBV status, immunosuppression regimen, and transplanted organ (5, 29, 30). The incidence of PTLD in LTx patients varies in the literature from 6 to 10% (12, 24). Montone et al. (23) reported a very high incidence of 20% that seems to be an outlier. It is of note that the cohort of transplant patients in that series was rather small (45 patients). A recent series by Levine and colleagues (22; 109 transplant patients) reported a lower PTLD incidence (1.8%) and mortality (1%) in their LTx patients compared with the case in previous reports. This was attributed to initial use of gancyclovir, followed by prolonged use of acyclovir at their institution (22). The incidence of PTLD in our cohort of 244 LTx patients, though slightly higher (3.3%), is comparable to that in this latter study (22). Prolonged use of antiviral therapy is not part of the treatment protocol for transplant patients at our institution, and the lower incidence may be a reflection of improved clinical management and close follow-up of these patients. Despite this, PTLD still appears to be an important cause of morbidity and mortality in LTx patients, behind rejection and infection, as reported elsewhere (12).
Several characteristics of the LTx PTLDs appeared to stand out when compared with our experience with those in other solid organ recipients. First, there was a high incidence of PTLDs occurring in the transplanted organ (62.5%). This high rate of lung-localized PTLDs in our patients is also significantly higher than the 18% suggested in the literature (31), though there have been reports indicating a similar high incidence (60–80%; 11, 32). Several explanations for this have been offered in the LTx setting (32). It has been postulated that there may be inoculation of bronchial lymphoid tissue by EBV from the oropharynx of the patient via aspiration. The donor lung may also be a direct source of infection as both the bronchial lymphoid tissue and donor lymphocytes may be a reservoir of EBV DNA. Finally, the relatively large amount of lymphoid tissue normally present in the transplanted lung may also increase the risk of PTLD. From this, one might expect the PTLD to be of donor origin. Although most PTLDs in solid organ transplant patients have been shown to be of host origin (33, 34), PTLDs in the allograft organ have been shown to be of donor origin (35). There is some difference of opinion in the literature because some authors have demonstrated that they can be of host origin as well (36). Analysis of origin of the PTLD was not done in this study, nor has it been studied extensively in PTLDs in LTx patients.
Secondly, PTLDs in LTx patients appear to occur very early after transplant (median time 7 mo) compared with non-LTx patients (median time, 41 mo), which is consistent with the case in the previous literature (12, 23, 24, 30). In addition, these PTLDs also appear to be clinically aggressive, with 5 of 8 patients dying of their PTLD. Kaplan-Meier analysis showed LTx patients to have a significantly shorter survival time compared with other solid organ PTLDs. The reasons for these differences are uncertain; however, factors to consider include host factors such as age and disease stage, as well as immunosuppression regimens.
Age and disease stage do not appear to account for this difference. Although our LTx patients were slightly older than were other organ recipients, there was no significant difference in mean age among the different groups. It is also unlikely that stage independently accounts for the poor performance of the former because there was no difference in stage of disease between the two groups. The degree of immunosuppression might also play a role in the early occurrence of PTLD because it is a well-known risk factor for PTLD (31). Original reports of PTLDs in solid organ recipients reported a relatively long interval to PTLD (2); however, with the use of Cyclosporine (CSA) and OKT3, this interval has markedly decreased (37). Although immunosuppression is somewhat individualized, guidelines for immunosuppression are not substantially different between the different organs in our institution. However, the dosage of CSA and steroids in our protocols is higher in LTx patients when compared with other solid organ Tx recipients (Table 5) and may be, in part, to blame. Anti-lymphocyte therapy is not an issue in this patient population because it was not part of their immunosuppression regimen. Although we were unable to specifically examine individual dosages, Levine et al. (22) reported that the immunosuppression regimens between LTx patients who developed PTLD and the remainder of the LTx population were not substantially different. Finally, although treatment was not uniform, some clinicians attempted to decrease immunosuppression before treatment with other agents. However, clinical circumstances may not allow significant reduction for fear of rejection. This limitation might also help explain the aggressive behavior of these PTLDs.
Third, monoclonality of LTx PTLDs was the rule, with all tested cases demonstrating a monoclonal B-lymphoid population. When Southern blot analysis was done on the LTx PTLD, these tumors demonstrated a strongly hybridizing rearranged Ig heavy chain gene band. This, as Locker and Nalesnik observed, correlated with the tumor containing a large proportion of neoplastic cells (14). A dominant band on SBA was associated with a poor response to reduction in immunosuppression and a worse prognosis when compared with patients who had weak bands (14). The poor clinical outcome in our patients is consistent with this observation.
Patient 8 is worthy of separate consideration. Multiple tumors developed simultaneously in the same site (colon) in this patient. Morphologically they were heterogeneous and molecular testing demonstrated that many of them were probably independent tumors, as indicated by different B-cell rearrangement patterns by SBA. Thus, as previous studies reported, multiple PTLDs occurring in the same patient can be of varying clonal origin (36, 38, 39). This may have clinical implications, since different clones may behave differently during therapy.
In summary, we present our findings in a series of PTLDs in LTx patients seen at our institution. To the best of our knowledge, this is the largest series of LTx patients reported. The incidence of PTLDs in LTx patients in our series is 3.3%. They are generally EBV-associated B-cell lymphoproliferative disorders that are monoclonal. Lung PTLDs in this series were most often monomorphic with 5 of the 8 (63%) patients presenting initially with this morphologic subtype. They have a propensity to occur early after transplantation and involve the transplanted organ, and survival is short. Their aggressive clinical course and early presentation after transplantation may be due to the relatively high doses of immunosuppression used in LTx recipients and limited options in decreasing it. Careful monitoring of these patients is warranted because of the poor prognosis of those who develop this complication of transplantation.
References
McKhann CF . Primary malignancy in patients undergoing immunosuppression for renal transplantation. Transplantation 1969; 8(2): 209–212.
Frizzera G, Hanto DW, Gajl-Peczalska KJ, Rosai J, McKenna RW, Sibley RK, et al. Polymorphic diffuse B-cell hyperplasias and lymphomas in renal transplant recipients. Cancer Res 1981; 41(11 Pt 1): 4262–4279.
Nalesnik MA, Jaffe R, Starzl TE, Demetris AJ, Porter K, Burnham JA, et al. The pathology of posttransplant lymphoproliferative disorders occurring in the setting of cyclosporine A-prednisone immunosuppression. Am J Pathol 1988; 133: 173–192.
Nalesnik MA, Makowka L, Starzl TE . The diagnosis and treatment of posttransplant lymphoproliferative disorders. Curr Probl Surg 1988; 25: 367–472.
Swerdlow SH . Post-transplant lymphoproliferative disorders: a morphologic, phenotypic and genotypic spectrum of disease. Histopathology 1992; 20: 373–385.
Swerdlow SH . Classification of the posttransplant lymphoproliferative disorders: from the past to the present. Semin Diagn Pathol 1997; 14: 2–7.
Liebowitz D . Epstein-Barr virus and a cellular signaling pathway in lymphomas from immunosuppressed patients. N Engl J Med 1998; 338: 1413–1421.
Hanto DW, Sakamoto K, Purtilo DT, Simmons RL, Najarian JS . The Epstein-Barr virus in the pathogenesis of posttransplant lymphoproliferative disorders. Clinical, pathologic, and virologic correlation. Surgery 1981; 90: 204–213.
Cleary ML, Nalesnik MA, Shearer WT, Sklar J . Clonal analysis of transplant-associated lymphoproliferations based on the structure of the genomic termini of the Epstein-Barr virus. Blood 1988; 72: 349–352.
Saemundsen AK, Klein G, Cleary M, Warnke R . Epstein-Barr-virus-carrying lymphoma in cardiac transplant recipient. Lancet 1982; 2(8290): 158.
Nalesnik MA . Clinical and pathological features of post-transplant lymphoproliferative disorders (PTLD). Springer Semin Immunopathol 1998; 20: 325–342.
Armitage JM, Kormos RL, Stuart RS, Fricker FJ, Griffith BP, Nalesnik M, et al. Posttransplant lymphoproliferative disease in thoracic organ transplant patients: ten years of cyclosporine-based immunosuppression. J Heart Lung Transplant 1991; 10: 877–886.
Locker J, Nalesnik M . Molecular genetic analysis of lymphoid tumors arising after organ transplantation. Am J Pathol 1989; 135: 977–987.
Nalesnik MA, Locker J, Jaffe R, Demetris AJ, Hartle K, Burnham JA, et al. Clonal characteristics of posttransplant lymphoproliferative disorders. Transplant Proc 1988; 20(1 Suppl 1): 280–283.
Knowles DM, Cesarman E, Chadburn A, Frizzera G, Chen J, Rose EA, et al. Correlative morphologic and molecular genetic analysis demonstrates three distinct categories of posttransplantation lymphoproliferative disorders. Blood 1995; 85: 552–565.
Cesarman E, Chadburn A, Liu YF, Migliazza A, Dalla-Favera R, Knowles DM . BCL-6 gene mutations in posttransplantation lymphoproliferative disorders predict response to therapy and clinical outcome. Blood 1998; 92: 2294–2302.
Niedobitek G, Mutimer DJ, Williams A, Whitehead L, Wilson P, Rooney N, et al. Epstein-Barr virus infection and malignant lymphomas in liver transplant recipients. Int J Cancer 1997; 73: 514–520.
Caillard S, Lachat V, Moulin B . Posttransplant lymphoproliferative disorders in renal allograft recipients: report of 53 cases of a French multicenter study. PTLD French Working Group. Transpl Int 2000; 13(Suppl 1): S388–S393.
Cockfield SM, Preiksaitis JK, Jewell LD, Parfrey NA . Post-transplant lymphoproliferative disorder in renal allograft recipients. Clinical experience and risk factor analysis in a single center. Transplantation 1993; 56(1): 88–96.
Rizell M, Olausson M, Friman S . PTLD in liver transplant recipients. Transplant Proc 2001; 33(4): 2455.
So S . CMV and EBV-PTLD after liver transplantation. Transplant Proc 2001; 33(1–2): 1317–1319.
Levine SM, Angel L, Anzueto A, Susanto I, Peters JI, Sako EY, et al. A low incidence of posttransplant lymphoproliferative disorder in 109 lung transplant recipients. Chest 1999; 116: 1273–1277.
Montone KT, Litzky LA, Wurster A, Kaiser L, Bavaria J, Kotloff R, et al. Analysis of Epstein-Barr virus-associated posttransplantation lymphoproliferative disorder after lung transplantation. Surgery 1996; 119: 544–551.
Aris RM, Maia DM, Neuringer IP, Gott K, Kiley S, Gertis K, et al. Post-transplantation lymphoproliferative disorder in the Epstein-Barr virus-naive lung transplant recipient. Am J Respir Crit Care Med 1996; 154: 1712–1717.
Schumann KW, Oriba HA, Bergfeld WF, Hsi ED, Hollandsworth K . Cutaneous presentation of posttransplant lymphoproliferative disorder. J Am Acad Dermatol 2000; 42(5 Pt 2): 923–926.
Sreenan JJ, Pettay JD, Tbakhi A, Totos G, Sandhaus LM, Miller ML, et al. The use of amplified variable number of tandem repeats (VNTR) in the detection of chimerism following bone marrow transplantation. A comparison with restriction fragment length polymorphism (RFLP) by Southern blotting. Am J Clin Pathol 1997; 107: 292–298.
Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, et al. The World Health Organization classification of neoplastic diseases of the haematopoietic and lymphoid tissues: report of the Clinical Advisory Committee Meeting, Airlie House, Virginia, November 1997. Histopathology 2000; 36(1): 69–86.
Gulley ML . Molecular diagnosis of Epstein-Barr virus-related diseases. J Mol Diagn 2001; 3: 1–10.
Penn I . Post-transplant malignancy: the role of immunosuppression. Drug Saf 2000; 23: 101–113.
Walker RC, Marshall WF, Strickler JG, Wiesner RH, Velosa JA, Habermann TM, et al. Pretransplantation assessment of the risk of lymphoproliferative disorder. Clin Infect Dis 1995; 20: 1346–1353.
Penn I . Cancers complicating organ transplantation. N Engl J Med 1990; 323: 1767–1769.
Yousem SA, Randhawa P, Locker J, Paradis IL, Dauber JA, Griffith BP, et al. Posttransplant lymphoproliferative disorders in heart-lung transplant recipients: primary presentation in the allograft. Hum Pathol 1989; 20: 361–369.
Maeda K, Hawkins ET, Oh HK, Kini SR, Van Dyke DL . Malignant lymphoma in transplanted renal pelvis. Arch Pathol Lab Med 1986; 110: 626–629.
Penn I . Host origin of lymphomas in organ transplant recipients. Transplantation 1979; 27(3): 214.
Spiro IJ, Yandell DW, Li C, Saini S, Ferry J, Powelson J, et al. Brief report: lymphoma of donor origin occurring in the porta hepatis of a transplanted liver. N.Engl J Med 1993; 329: 27–29.
Chadburn A, Cesarman E, Liu YF, Addonizio L, Hsu D, Michler RE, et al. Molecular genetic analysis demonstrates that multiple posttransplantation lymphoproliferative disorders occurring in one anatomic site in a single patient represent distinct primary lymphoid neoplasms. Cancer 1995; 75: 2747–2756.
Swinnen LJ, Fisher RI . OKT3 monoclonal antibodies induce interleukin-6 and interleukin-10: a possible cause of lymphoproliferative disorders associated with transplantation. Curr Opin Nephrol Hypertens 1993; 2(4): 670–678.
Kaplan MA, Ferry JA, Harris NL, Jacobson JO . Clonal analysis of posttransplant lymphoproliferative disorders, using both episomal Epstein-Barr virus and immunoglobulin genes as markers. Am J Clin Pathol 1994; 101: 590–596.
Cleary ML, Sklar J . Lymphoproliferative disorders in cardiac transplant recipients are multiclonal lymphomas. Lancet 1984; 2(8401): 489–493.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ramalingam, P., Rybicki, L., Smith, M. et al. Posttransplant Lymphoproliferative Disorders in Lung Transplant Patients: The Cleveland Clinic Experience. Mod Pathol 15, 647–656 (2002). https://doi.org/10.1038/modpathol.3880581
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880581
This article is cited by
-
Immundefizienz und Immunsuppression
Der Pathologe (2011)
-
Transplantationsassoziierte Pathologie der Lunge
Der Pneumologe (2011)
-
Gastrointestinal Involvement of Posttransplant Lymphoproliferative Disorder in Lung Transplant Recipients: Report of a Case
Diseases of the Colon & Rectum (2005)
