
Abstract
Gastrointestinal stromal tumors (GISTs) coexpress CD34 and the Kit tyrosine-kinase receptor (CD117). A subset of GISTs carry gain-of-function mutations of the c-kit proto-oncogene in its juxtamembrane domain. The relationship between the mutational status and histological as well as immunohistochemical features has not been assessed in detail. 36 GISTs and 14 other gastrointestinal mesenchymal tumors were investigated for their morphology and immunophenotype as well as for the presence of c-kit mutations. DNA was extracted from formalin-fixed, paraffin-embedded tissue. Exons 9, 11, 13, and 17 of c-kit were analyzed by SSCP. Bands with altered mobility were excised, reamplified, and sequenced. C-kit mutations in Exon 11 encoding the juxtamembrane domain were identified in 19 cases (52.8%), with deletions in 12 cases, insertions in 3 cases (2 of these as duplications), and point mutations in 4 cases. The mutations clustered between Codons 553 and 561, pinpointing the critical region for deregulated Kit receptor activation. In both Exons 9 and 13, single mutations could be identified, whereas no mutations were found in Exon 17. There were c-kit mutations in 66.6% of benign GISTs (14/21), 83.3% of the malignant (5/6), and 40% of the cases of intermediate malignancy (2/5). A low frequency of mutations in benign GISTs, as reported previously by other researchers, could not be observed in our panel. Interestingly, all GISTs with c-kit mutations displayed a spindle cell phenotype, whereas mutations were absent in all 7 tumors with an epithelioid component (P = .03). This finding suggests a relationship between c-kit mutation and histological subtype in GISTs.
Similar content being viewed by others

INTRODUCTION
The value of immunohistochemical markers in the diagnosis of gastrointestinal stromal tumors (GISTs) is well established (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11). A high percentage of GISTs expresses CD34, BCL-2, and vimentin, in contrast to the cases of leiomyomas and Schwann cell tumors. The vast majority of GISTs investigated up to now additionally express CD117, the transmembrane growth factor receptor for stem cell factor (SCF) Kit. The Kit protein is a Type III receptor tyrosine kinase and is important for the development and survival of mast cells, hematopoetic stem cells, melanocytes, germ cells, and certain cutaneous epithelial cells. A coexpression of the Kit receptor with CD34 has been observed by several groups in the interstitial cells of Cajal (ICCs) in the gastrointestinal tract, which are considered by some authors to be potential cells of origin of GISTs (11, 12, 13). Further insight into the role of the Kit receptor for the pathogenesis of GISTs was gained by the identification of gain-of-function mutations in the c-kit proto-oncogene in a subset of GISTs (14, 15, 16). Recently, an association of the presence of c-kit mutations and poor prognosis was suggested, whereas no correlations were found with either the mitotic index or the tumor size (17). In another series, mutations of c-kit were found preferentially in malignant GISTs (18). The relationship between the mutational status and morphological features, however, has not yet been clarified. In the present study, we investigated 36 GISTs for these parameters in comparison with 14 other mesenchymal tumors. We compared our results with those in the literature with special reference to frequency and localization of c-kit mutations and their correlation with morphological features.
MATERIALS AND METHODS
Tissues
Thirty-six cases of GISTs were examined histologically and immunohistochemically. Of these, 31 cases were obtained from the Department of Pathology, University of Bonn Medical Center, and 5 tumors had been submitted by the Institute of Pathology, Essen, Germany. All tissues had been fixed in 4% neutral buffered formalin and routinely processed into paraffin.
The GISTs and the other gastrointestinal tumor entities were diagnosed according to the criteria of the current World Health Organization classification (19). Using a modified grading system according to Franquemont et al. (20), these tumors were classified as benign, malignant, or of intermediate (uncertain) malignancy. Tumors with >5 mitoses per 50 HPF and an MIB-1 index of ≥5% were classified as malignant; cases with >2 mitotic figures per 50 HPF and a MIB-1 index of ≤2% were graded as benign; all other cases were graded as GISTs of intermediate (uncertain) malignancy. The present panel thus consisted of 25 benign, 6 malignant, and 5 GISTs of intermediate malignancy.
Control tissues included normal gastric, small intestinal, and colonic tissue samples. Nontumorous tissue adjacent to the tumors served as internal control. Additionally, 14 other mesenchymal tumors of the gastrointestinal tract were investigated, specifically, 10 leiomyomas, 1 leiomyosarcoma, and 3 Schwann cell tumors.
Morphological andImmunohistochemical Studies
For morphological evaluation and mitotic counts, all cases were stained with hematoxylin and eosin. The immunohistochemical methods, antibodies, dilutions, and sources are summarized in Table 1. Studies on serial sections (4 μm) were performed by using the avidin-biotin-peroxidase complex detection system with diaminobenzidine tetrahydrochloride (for CD117) or 3-amino-9-ethyl-carbazole (for the other antibodies) as chromogens.
The mitotic index was determined by counting 50 high-power fields (HPFs, 400×). In some smaller biopsies, in which 50 HPFs could not be evaluated, the results were multiplied by 50 and divided by the number of fields counted to obtain comparable results. The Ki-67 score was determined by counting a minimum of 1000 tumor cells and was expressed as the percentage of positive nuclei. Immunohistochemical results were assessed in a semiquantitative manner using the following categories: intensity (3, strong; 2, intermediate; 1, weak; and —, none) and fraction of positive cells (+++, >75%; ++, 75–10%; +, <10%; and —, none).
DNA Extraction
Serial sections (10 μm) of the paraffin blocks were deparaffinized by xylene. Total DNA was extracted after pretreatment with proteinase K and absorption on silica gel membranes (Qiagen, Hilden, Germany) and analyzed by single-strand conformational polymorphism analysis (SSCP). Therefore, intronic PCR primers were designed to amplify Exons 9, 11, 13, and 17 (Table 2). PCR was performed in 10-μL reactions containing 1.0 μL DNA, 10 mm Tris-HCl (pH 8.3), 40 mm KCl, 1.0–1.5 mm MgCl2, 200 mm of each dNTP, 20 pm of each primer, and 0.25 U Platinum Taq polymerase (Life Technologies, Invitrogen GmbH, Karlsruhe, Germany). PCR reaction was carried out on an Uno II Thermoblock (Biometra, Göttingen, Germany). Initial denaturation at 94° C for 3 minutes was followed by 41 cycles and a final extension step (5 min at 72° C). The cycles included denaturation at 94° C for 40 seconds, annealing at 55–57° C for 40 seconds, and extension at 72° C for 35 seconds. PCR products were diluted with formamide and denaturated at 94° C for 10 minutes, and single strands were separated on polyacrylamide gels under two different conditions (Table 3). Single and double strands of the PCR products were visualized by silver staining as described elsewhere (21). DNA single-strand bands showing an altered mobility in comparison to reference products were excised from the wet gel. DNA was eluted in H2O for 2 hours at 50° C, precipitated by centrifugation at 12,000 × g for 30 minutes, and reamplified. The products were purified using spin columns (QIAquick PCR Purification Kit, Qiagen). Cycle sequencing (ABI PRISM Dye Terminator Sequencing Ready Reaction Kit, Applied Biosystems, Weiterstadt, Germany) was done on a TC 9600 thermocycler (Perkin Elmer, Rodgau-Jügesheim, Germany) with 20 ng of PCR product as template according to the protocol of the manufacturer. The sequencing products were separated on a 6%, 1:19 bisacrylamide:acrylamide gel on an ABI 373A sequencer (Applied Biosystems). All sequence alterations were confirmed by an independent PCR amplification, followed by SSCP, reamplification, and sequencing to exclude PCR artifacts.
Statistics
Significance of association of the presence of c-kit mutations with clinical and histological parameters was tested by Fisher’s Exact test and by univariate (ANOVA) and multivariate analysis of covariance (MANCOVA), as indicated.
RESULTS
Clinical Data
The clinical data are summarized in Table 4. Seventeen male and 19 female patients with GISTs were included. There was no statistical significant difference between sex distribution or age at diagnosis between GISTs with spindled cells and epithelioid component (mean, 66.7 vs 65.7 y).
For 21 of 36 patients, data on the clinical follow-up were available. Four patients with incidental benign GISTs died because of a synchronous malignancy (three cases of gastric carcinoma, one pancreatic carcinoma). One patient had liver metastases of the GIST at the time of diagnosis. Another two patients developed liver metastases during follow-up (12 and 29 mo after the initial diagnosis, respectively). 14 patients (including 1 malignant GIST and 2 tumors classified as of intermediate malignancy) were free of disease during a follow-up period ranging from 26 to 94 months (mean, 52.9 mo).
Macroscopy and Light Microscopy
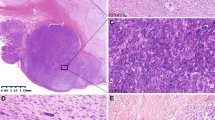
Of the 36 GISTs, 29 were predominantly of the spindle cell type (81%; Fig. 1A), 3 were predominantly epithelioid (8%; Fig. 1B), and 4 (11%) were of the mixed type, consisting of spindle and epithelioid cells (the latter component ranging between 30 and 80%). Of the seven epithelioid and mixed GISTs, three tumors were classified as benign, three cases as malignant, and one tumor as of intermediate malignancy. The diameter of the smallest tumor measured 0.1 cm; that of the largest, 23 cm. All cases considered nonbenign measured >5 cm in diameter.

A–H, morphological and immunohistochemical features of a spindle cell (A, C, E, G) and an epithelioid (B, D, F, H) gastrointestinal stromal tumor. A, the spindle cells are arranged in storiform and herringbone patterns and have blunt-ended nuclei. B, the epithelioid cells are arranged in organoid clusters and have round nuclei and abundant cytoplasm. Both subtypes express CD34 (C and D), bcl-2 (E and F), and the Kit receptor (G and H). Magnifications, 240×.
Immunohistochemistry
The immunohistochemical results are summarized in Table 5. The Kit receptor was expressed in the vast majority of tumor cells of all cases. Only one GIST showed an intermediate expression in <75% of tumor cells. CD34 was expressed in 26 GISTs in >75% of the tumor cells; another 7 cases showed a CD34 expression in <75%. Three tumors stained strongly in <10% of the cells. None of the cases was completely negative for CD34 or Kit. Bcl-2 and vimentin were found to be variably expressed. In four cases, one of both markers was completely absent; another case showed neither bcl-2 nor vimentin expression. None of the 36 GISTs showed an intensive expression of α-smooth muscle actin (SMA), desmin, or protein S-100 in the majority of tumor cells, whereas internal control structures (such as the muscularis propria and small peripheral nerves) showed a distinct positive reaction. There were no significant differences between spindle cell and epithelioid tumors concerning the immunohistochemical phenotype (Fig. 1C–H).
None of the other mesenchymal tumors expressed the Kit receptor. Positive mast cells served as internal control. In some leiomyomas, scattered CD117-positive spindle cells were detected between the smooth muscle bundles and, because of their coexpression of CD34, were interpreted as ICCs. In one leiomyoma, small foci of CD34-positive cells were observed; in the other leiomyomas and Schwann cell tumors, CD34 expression was found only in vascular structures. From the other mesenchymal tumors, one Schwann cell tumor showed an intermediate bcl-2 expression in most of the tumor cells. All Schwann cell tumors strongly expressed vimentin and S-100, but none of the leiomyomas did. All leiomyomas strongly expressed SMA and desmin; the leiomyosarcoma, only SMA.
c-kit Mutations
DNA was obtained from 36 GISTs and from 14 other mesenchymal tumors. Of the GISTs, 19 (52.8%) showed mutations of c-kit in Exon 11 (see Fig. 2). Deletions (between 3 and 45 bp in length) were found in 12 cases, point mutations in 4 cases, and insertions in 3 cases (in one case combined with a point mutation in the second allele; in the other 2 cases, duplications). Seven of these mutations were novel mutations that have not been described before. In 74% of cases (14 of 19), the mutations in Exon 11 clustered in the region between Codons 555 and 560 (for examples, see Fig. 3). Five other mutations in Exon 11 were located outside this region, with one 3-bp deletion at Codon 579, one 21 bp-deletion at Codon 570, one point mutation at Codon 576, one 6 bp-insertion at Codon 577 (duplication) and another 27 bp-insertion at Codon 584 (duplication). All cases were in-frame mutations. Single mutations were found in Exon 9 (Fig. 4, Case 41) and Exon 13 (Fig. 4, Case 47), the first being a 6-bp insertion at Codon 504 (duplication); the latter, a point mutation at Codon 642. Of the 21 cases of benign GISTs, 14 had mutations (66.6%), 5 additional mutations were found in 6 malignant (83.3%) and 2 mutations in 5 cases of intermediate malignancy (40%). The GISTs with mutations showed exclusively a spindled phenotype. None of tumors with epithelioid component showed mutations in Exon 9, 11, 13, or 17. All non-GIST mesenchymal tumors lacked any mutations in Exon 9, 11, 13, or 17 of c-kit.

Predicted amino acid sequences in Exons 9, 11, and 13. Numbers shown above the wild-type amino acid sequence indicate codons. Point mutations and changed amino acids are indicated in an empty square, insertions in a black square. Deleted amino acids are shown by dashes (—).

Single-strand conformational polymorphism analysis and sequence of Exon 11 in GISTs. Case 1 carries a 3-bp deletion corresponding to the deletion of Asp-579. The point mutation in Case 6 leads to the replacement of Val-559 by Asp. In Case 7, Trp-557 and Lys-558 are deleted; in Case 11, a deletion of 9 bp and concurrent insertion of 3 bp leads to the replacement of Gln-556, Trp-557, Lys-558, and Val-559 by His and Thr. In Case 10, two amino acids (Gln and Leu) are inserted at Codon 577.

Single-strand conformational polymorphism analysis and sequence analysis of Exons 9 and 13 in gastrointestinal stromal tumors. Case 41: Exon 9 shows a duplication of six nucleotides encoding for Ala-502 and Tyr-503. Case 47: In Exon 13, the mutation of A to G leads to the substitution of Lys-642 by Glu.
C-kit mutational status was correlated with the clinicopathological findings (Table 5). C-kit mutations were found to be associated with the MIB-1 index (P = .031; univariate analysis, ANOVA). Multivariate analysis of covariance (MANCOVA) showed that the presence of c-kit mutations was an independent parameter associated with elevated MIB-1 (P = .01). The presence of c-kit mutations was also associated with histology; using Fisher’s Exact test, c-kit mutations were associated with spindled morphology of the GISTs (P = .03).
DISCUSSION
Gastrointestinal stromal tumors are the most common subset of mesenchymal tumors of the gastrointestinal tract (22). Their diagnosis by histological and immunohistochemical criteria is well established (1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 19). The best marker to distinguish these tumors from leiomyomas and Schwann cell tumors is the Kit receptor (CD117; 11). Other helpful markers, especially when used in combination, are CD34 (4), bcl-2, and vimentin.
The question of the cell of origin of GISTs has been discussed by several groups. Kindblom et al. (12) strongly suggested that GISTs originate from ICCs or from a common stem cell of ICC and smooth muscle cells. The detection of the embryonic form of smooth-muscle myosin heavy chain (Smemb/MHC-B) in GISTs and ICCs (23) supports this theory. However, recent studies of Vanderwinden et al. (24) question the coexpression of CD34 and the Kit receptor in ICCs. They suggest that although ICCs express CD117, CD34 is found in directly adjacent fibroblastic cells in the tunica muscularis but not in the ICCs themselves. The fact that a group of extragastrointestinal mesenchymal tumors, namely omental and mesenteric neoplasms, share morphological features as well as the coexpression of CD34 and Kit with GISTs suggests a primitive precursor cell existing in and outside the gastrointestinal tract that could give rise to GISTs (6). On the other hand, Robinson et al. (13) recently confirmed the possibility of a subset of ICCs coexpressing CD34 and the Kit receptor. By using single-cell PCR on isolated ICCs obtained from a mouse model and sequential immunohistochemical staining on the same section from the human small intestine, they demonstrated that there are three different spindle cell populations surrounding Auerbach’s plexus: a majority of ICCs only positive for Kit, another cell population only expressing CD34, and a minority of ICCs coexpressing CD34 and Kit. The latter type could be the progenitor cells of GISTs.
Recently, gain-of-function mutations of the c-kit gene were described in a subset of GISTs (14, 15, 16). Several studies suggested a possible role of these mutations in biological behavior and prognosis (3, 17, 25). Lasota et al. (18) found mutations in Exon 11 of c-kit encoding the juxtamembrane region in the majority of malignant GISTs (12 of 24), but only in one benign tumor (1 of 19). They concluded that c-kit mutations occur preferentially in malignant GISTs and might be a clinically useful marker in their evaluation. In contrast, Sakurai et al. (26) could not confirm that c-kit mutations are always related to poor prognosis. In our panel of GISTs, there was a tendency towards a higher mitotic count, but this was not statistically significant. However, when we analyzed the association of MIB-1 index, we found a significant higher MIB-1 index in GISTs carrying c-kit mutations (P = .01, MANCOVA). This suggests that the presence of constitutively active c-Kit receptors may result in an increased proliferation activity in GIST cells. The overall frequency of c-kit mutations detected in our study (21 of 36 cases, 58%) was considerable higher than the rate described in other studies (for example, 20% by Moskaluk et al.; 25). Possible explanations may be a higher detection rate by the methods that we used or a geographical difference of mutation rates between different populations. However, the initial study of Hirota (14) found 5 mutations in 6 cases (83%). We also found a rather high rate of mutations in malignant GISTs (5 of 6, 83%), whereas the frequency of mutations in benign GISTs of 67% in our study (14 of 21) was significantly higher than that observed in previous studies (14, 17, 18). Possible explanations for the differences in the frequency of c-kit mutations in benign and malignant GISTs in several studies may be different proportions of benign and malignant cases in these series or differences in classification.
Mutations could be detected in our study already in clear-cut benign lesions as small as 0.6 cm in largest diameter. This argues against the possibility that c-kit mutations may occur as a late event during progression of the tumors. In addition, 7 out of 17 GISTs measuring >5 cm in largest diameter did not show mutations of c-kit. These observations confirm the results from Ernst et al. (17), who could not detect a correlation of c-kit mutations with tumor size. DeMatteo et al. (2) postulated tumor size to be one of the most reliable predictors of tumor prognosis. The fact that mutations occur both in large and small tumors indicates that c-kit mutation may not be a useful predictor for poor outcome.
In contrast, our study indicates that the occurrence of c-kit mutations is associated with specific histological features. No mutations were detectable in seven GISTs with a significant epithelioid component. This group consisted of predominantly epithelioid and mixed epithelioid and spindle cell GISTs. The difference of frequency of c-kit mutations between the histological subtypes was significant (P = .03, Fisher’s Exact test). The only other study on c-kit mutations (18) that mentioned the histological phenotypes of the tumors found a single mutation in eight epithelioid cases (12.5%), in contrast to 15 mutations in 35 spindle cell GISTs (42%). This observation strongly suggests a relationship of pheno- and genotype in GISTs and an involvement of c-kit mutations in the pathogenesis of spindle cell GISTs. In our study, 21/29 spindle cell type GISTs (74%) carried mutations of the c-kit gene. This does not exclude an ever higher mutation rate in this subtype because individual mutations may be missed by the SSCP screening method or may be located outside the mutational hot spot regions screened in this study. The results indicate that c-kit is activated by mutations in the majority of spindle cell type GISTs. In contrast, GISTs with a significant epithelioid component may be characterized by other pathogenetic alterations that have not been identified yet. However, a recent study of Lasota et al. (27) indicated that the epithelioid subtype may also harbor c-kit mutations outside Exon 11.
Another important question of our study concerns the localization and type of mutations as well as their possible functional role. A summary of most published cases up to now (7, 14, 16, 17, 18, 25, 26, 28) and our own findings (together, 150 cases) shows that all tumors had in-frame mutations (see Fig. 5). More than three quarters of the cases had deletions; approximately one fifth of cases had simple changes of one or two amino acids. All mutations in Exon 11 were located between residues 550 and 592 located in the juxtamembrane region of c-kit (see Fig. 6). Of all published 150 GISTs with mutations, 128 tumors (85%) showed an involvement of the region between 553 and 561 with different types of mutation. In our own study, 14 of 19 cases (74%) showed an involvement of this region in Exon 11. The clustering of mutations in this specific region suggests that the mutations may be gain-of-function mutations, either by generating a new mechanism leading to increased signaling or by disrupting a receptor site that normally has a negative function on the receptor activity. First insights on potential interesting residues in the proto-oncogene c-kit were given by Herbst et al. (29). He and his coworkers generated mutations in the feline homologue of c-kit and identified inhibitory as well as transforming mutations. Most interestingly, deletions of tyrosine-569 and valine-570 (corresponding to human tyr568 and val569) resulted in an increased transforming and mitogenic activity of feline c-kit. In fact, deletions of this region were also found in gastrointestinal stromal tumors, suggesting a similar mechanism of activation. A possible mechanism by which mutations in the juxtamembrane region may lead to an activation of the Kit receptor is the disruption of a receptor site for a negatively regulating molecule. This could be the case for the SHP-1 tyrosine phosphatase, which binds to tyrosine 570 (30). SHP-1 is capable of negatively regulating a broad spectrum of cytokine signaling pathways, including the SCF/Kit signaling pathway. Kozlowski and coworkers (30) were able to show that a mutation of the tyrosine 570 residue not only prevented the binding of SHP-1 to the Kit receptor but also resulted in a hyperproliferative response to SCF. However, the SHP-1 function has not been studied in detail in GIST cells.

A review of mutations in Exon 11 in 150 cases published until now. On the right side, number of cases per mutation is indicated; under ref., authors are symbolized as follows: *Ernst et al. (4), +Hirota et al. (9), <Lasota et al. (14), =Miettinen et al. (24), ⁁Moskaluk et al. (28), Ñakahara et al. 1998 (29), $Sakurai et al. 1999 (31), §Taniguchi et al. 1999 (28), and >Wardelmann et al. 2002. The predominant type of mutation is a simple deletion of 1 to 15 amino acids.

Frequency of mutations per codon published until now (including our own cases). Each square symbolizes one case with an amino acid change or loss in this codon. A clustering of mutations is observed in the region between Codons 553 and 561 (128/150 cases, i.e., 85.3%).
The evaluation of all c-kit mutations in GISTs described so far suggests that a region adjacent but not identical to the possible docking side of SHP-1 may be the main target of mutational events: the region of the residues 553 to 561 was most frequently affected in GISTs (Fig. 3). Ma et al. identified this region as an amphipathic α-helix in the Kit intracellular juxtamembrane region. Mutagenesis of the residues Tyr-553, Trp-557, Val-559, or Val-560 (all located in this intracellular juxtamembrane region) resulted in a substantially increased spontaneous receptor phosphorylation (31). In normal cells, these amino acids therefore might prevent a spontaneous receptor phosphorylation and activation. The gain-of-function mutations first described by Hirota et al. (14) in GISTs were located in this region.
For the mutations distal of this region, the effect on the receptor function has to be clarified. Sakurai and coworkers (26) found that all patients with a deletion in the distal location had a good outcome. Further work has to be done to clarify the molecular events induced by mutations in this domain.
Lux et al. (32) found additional mutations in Exon 9 and 13 in eight tumors that lacked Exon 11 mutations. These were the first descriptions of mutations in c-kit exons encoding other regions than the juxtamembrane domain, i.e., the extracellular (Exon 9) and the kinase domain (Exon 13). Very recently, another group found identical mutations in Exons 9 and 13 (27). In our own 15 cases lacking mutations in Exon 11, we detected two additional cases with mutations in these two exons. Again, both tumors exhibited a spindle cell phenotype. Receptor activation in these mutations probably is a result of ligand-independent oligomerization (32). Lux et al. showed in a cell line established from one of their Exon 13 mutant GISTs that Kit is constitutively tyrosine phosphorylated in a ligand-independent manner. They postulated that the Exon 13 mutation may be activating by alteration of the three-dimensional structure of the mutant protein. For the extracellular domain mutations in Exon 9, Hirota et al. (33) showed very recently that the mutant Kit receptor is constitutively autophosphorylated in a ligand-independent manner, perhaps again via ligand-independent dimerization. However, the precise mechanism of constitutive activation has to be clarified.
In summary, our findings show that c-kit mutations occur preferentially in the spindle cell phenotype of GISTs. This suggests that GISTs are not a homogenous tumor entity but consist of at least two entities defined by molecular as well as histological characteristics. On the molecular level, a hot spot region between amino acids 553 and 561 could be identified by us and others. The functional study of this receptor region will elucidate the mechanism of oncogenic activation of c-kit.
References
Appelman HD . Mesenchymal tumors of the gastrointestinal tract.In: Ming SC, Goldman H, editors. Pathology of the gastrointestinal tract. Philadelphia: Saunders; 1992.p. 310–350.
DeMatteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF . Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors of survival. Ann Surg 2000; 231: 51–58.
Emory TS, Sobin LH, Lukes L, Lee DH, O'Leary TJ . Prognosis of gastrointestinal smooth-muscle (stromal) tumors. Dependence on anatomic site. Am J Surg Pathol 1999; 23: 82–87.
Miettinen M, Virolainen M, Sarlomo-Rikkala M . Gastrointestinal stromal tumors—value of CD34 antigen in their identification and separation from true leiomyomas and schwannomas. Am J Surg Pathol 1995; 19: 207–216.
Miettinen M, Sarlomo-Rikala M, Kovatich AJ . Cell-type and tumor-type related patterns of bcl-2 reactivity in mesenchymal cells and soft tissue tumors. Virchows Arch 1998; 433: 255–260.
Miettinen M, Monihan JM, Sarlomo-Rikala M, Kovatich AJ, Carr NJ, Emory TS, et al. Gastrointestinal stromal tumors/smooth muscle tumors (GISTs) primary in the omentum and mesentery: clinicopathological and immunohistochemical study of 26 cases. Am J Surg Pathol 1999; 23: 1109–1118.
Miettinen M, Sarlomo-Rikala M, Sobin LH, Lasota J . Esophageal stromal tumors: a clinicopathologic, immunohistochemical, and molecular genetic study of 17 cases and comparison with esophageal leiomyomas and leiomyosarcomas. Am J Surg Pathol 2000; 24: 211–222.
Miettinen M, Sarlomo-Rikala M, Sobin LH, Lasota J . Gastrointestinal stromal tumors and leiomyosarcomas in the colon. A clinicopathologic, immunohistochemical and molecular genetic study of 44 cases. Am J Surg Pathol 2000; 24: 1339–1352.
Miettinen M, Sobin LH, Sarlomo-Rikala M . Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with a reference to CD117. Mod Pathol 2000; 13(10): 1134–1142.
Miettinen M, Lasota J . Gastrointestinal stromal tumors—definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archiv 2001; 438: 1–12.
Sarlomo-Rikala M, Kovatich AJ, Barusevicius A, Miettinen M . CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol 1998; 11: 728–734.
Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM . Gastrointestinal pacemaker cell tumor (GIPACT)—gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 1998; 152: 1259–1269.
Robinson TL, Sircar K, Hewlett BR, Chorneyko K, Riddell RH, Huizinga JD . Gastrointestinal stromal tumors may originate from a subset of CD34-positive interstitial cells of Cajal. Am J Pathol 2000; 156: 1157–1163.
Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998; 279: 577–580.
Kitamura Y, Hirota S, Nishida T . Molecular pathology of c-kit proto-oncogene and development of gastrointestinal stromal tumors. Ann Chir Gynaecol 1998; 87: 282–286.
Nakahara M, Isozaki K, Hirota S, Miyagawa JI, Hase-Sawada N, Taniguchi M, et al. A novel gain-of-function mutation of c-kit gene in gastrointestinal stromal tumors. Gastroenterology 1998; 115: 1090–1095.
Ernst SI, Hubbs AE, Przygodzki RM, Emory TS, Sobin LH, O'Leary TJ . KIT mutation portends poor prognosis in gastrointestinal stromal/smooth muscle tumors. Lab Invest 1998; 78: 1633–1636.
Lasota J, Jasinski M, Sarlomo-Rikala M, Miettinen M . Mutations in exon 11 of c-kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol 1999; 154: 53–60.
Miettinen M, Blay JY, Sobin LH . Mesenchymal tumours of the stomach.In: Hamilton SR, Aaltonen LA, editors. World Health Organization classification of tumours. Pathology and genetics of tumours of the digestive system. 1st ed. Lyon, France: IARC Press; 2000: 62–65.
Franquemont DW . Differentiation and risk assessment of gastrointestinal stromal tumors. Am J Clin Pathol 1995; 103: 41–47.
von Deimling A, Bender B, Louis DN, Wiestler OD . A rapid and non-radioactive PCR based assay for the detection of allelic loss in human gliomas. Neuropathol Appl Neurobiol 1993; 19: 524–529.
Miettinen M, Sarlomo-Rikala M, Lasota J . Gastrointestinal stromal tumours. Ann Chir Gynaecol 1998; 87(4): 278–281.
Sakurai S, Fukasawa T, Chong JM, Tanaka A, Fukayama M . Embryonic form of smooth muscle myosin heavy chain (SMemb/MHC-B) in gastrointestinal stromal tumor and interstitial cells of Cajal. Am J Pathol 1999; 154: 23–28.
Vanderwinden JM, Rumessen JJ, De Laet MH, Vanderhaeghen JJ, Schiffmann SN . CD34+ cells in human intestine are fibroblasts adjacent to, but distinct from, interstitial cells of Cajal. Lab Invest 1999; 79: 59–65.
Moskaluk CA, Tian Q, Marshall CR, Rumpel CA, Franquemont DW, Frierson HF . Mutations of c-kit JM domain are found in a minority of human gastrointestinal stromal tumors. Oncogene 1999; 18: 1897–1902.
Sakurai S, Fukasawa T, Chong JM, Tanaka A, Fukayama M . C-kit gene abnormalities in gastrointestinal stromal tumors (tumors of interstitial cells of Cajal). Jpn J Cancer Res 1999; 90: 1321–1328.
Lasota J, Wozniak A, Sarlomo-Rikala M, Rys J, Kordek R, Nassar A, et al. Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. Am J Pathol 2000; 157: 1091–1095.
Taniguchi M, Nishida T, Hirota S, Isozaki K, Ito T, Nomura T, et al. Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res 1999; 59: 4297–4300.
Herbst R, Munemitsu S, Ullrich A . Oncogenic activation of v-kit involves deletion of a putative tyrosine-substrate interaction site. Oncogene 1995; 10: 369–379.
Kozlowski M, Larose L, Lee F, Le DM, Rottapel R, Siminovitch KA . SHP-1 binds and negatively modulates the c-Kit receptor by interaction with tyrosine 569 in the c-Kit juxtamembrane domain. Mol Cell Biol 1998; 18: 2089–2099.
Ma Y, Cunningham ME, Wang X, Ghosh I, Regan L, Longley BJ . Inhibition of spontaneous receptor phosphorylation by residues in a putative alpha-helix in the KIT intracellular juxtamembrane region. J Biol Chem 1999; 19: 13399–13402.
Lux ML, Rubin BP, Biase TL, Chen C-J, Maclure T, Demetri G, et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 2000; 156: 791–795.
Hirota S, Nishida T, Isozaki K, Taniguchi M, Nakamura J, Okazaki T, et al. Gain-of-function mutation at the extracellular domain of KIT in gastrointestinal stromal tumours. J Pathol 2001; 193: 505–510.
Acknowledgements
We thank Steffen Albrecht for critical reading of the manuscript as well as Inge Heim, Christiane Esch, Susanne Steiner, Barbara Reddemann, Dorota Denkhaus, and Gerrit Klemm for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wardelmann, E., Neidt, I., Bierhoff, E. et al. c-kit Mutations in Gastrointestinal Stromal Tumors Occur Preferentially in the Spindle Rather Than in the Epithelioid Cell Variant. Mod Pathol 15, 125–136 (2002). https://doi.org/10.1038/modpathol.3880504
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880504
Keywords
This article is cited by
-
Deep learning predicts patients outcome and mutations from digitized histology slides in gastrointestinal stromal tumor
npj Precision Oncology (2023)
-
The MAPK pathway functions as a redundant survival signal that reinforces the PI3K cascade in c-Kit mutant melanoma
Oncogene (2014)
-
DOG1 and PKC-θ are useful in the diagnosis of KIT-negative gastrointestinal stromal tumors
Modern Pathology (2011)
-
Brauchen wir in Deutschland Sarkomzentren?
Der Pathologe (2011)
-
qPCR in gastrointestinal stromal tumors: Evaluation of reference genes and expression analysis of KIT and the alternative receptor tyrosine kinases FLT3, CSF1-R, PDGFRB, MET and AXL
BMC Molecular Biology (2010)
