
Abstract
Whereas deep fibromatoses (abdominal, extra-abdominal, mesenteric) display locally aggressive behavior, superficial fibromatoses typically remain small and less likely to recur despite essentially identical morphology. Somatic β-catenin or APC gene mutations have been reported in ≤74% of sporadic deep fibromatoses and in virtually 100% of Gardner syndrome–associated fibromatoses, whereas genetic events in superficial fibromatoses remain less well characterized. We performed immunohistochemical staining for β-catenin on 29 superficial fibromatoses (22 palmar, 5 plantar, 1 penile, and 1 infantile digital fibromatosis) and 5 deep fibromatoses. Mutations of β-catenin and APC genes were analyzed in cases of superficial fibromatoses by direct DNA sequencing of the β-catenin gene on Exon 3 encompassing the GSK-3 36 phosphorylation region and of the APC gene on the mutation cluster region. Nuclear accumulation of β-catenin was present in 86% (25/29) of superficial fibromatosis cases ranging from 5 to 100% of nuclei (mean, 13%; median, 10%), though in a minority of nuclei in most examples. Deep fibromatoses had 60 to 100% nuclear staining in all five cases. No somatic mutations of β-catenin or APC genes were identified in any of the superficial fibromatoses. In contrast to deep fibromatoses, superficial fibromatoses lack β-catenin and APC gene mutations; the significance of focal nuclear β-catenin accumulation is unclear. This difference may account inpart for their divergent clinical manifestations despite their morphologic resemblance to deep fibromatoses.
Similar content being viewed by others

INTRODUCTION
The fibromatoses are classified as superficial and deep (1). The superficial fibromatoses encompass small lesions of the hands (palmar fibromatoses/Dupuytren's contractures, knuckle pads, infantile digital fibromatoses), feet (plantar fibromatoses/Ledderhose's disease), and penis (penile fibromatoses/Peyronie's disease; 2). Palmar fibromatosis is the most common of these, occurring in 1 to 2% of the population with a male predominance, frequently presenting bilaterally. Plantar fibromatoses occur in a younger age group and are also prone to bilaterality. As the name implies, infantile digital fibromatoses arise in the digits of infants and young children and are characterized by peculiar inclusion-like condensations of cytoplasmic actin (3). Penile fibromatoses are uncommon and tend to present in association with other superficial fibromatoses. The deep fibromatoses (aggressive fibromatoses, musculoaponeurotic fibromatoses, desmoid tumors) are rarer, encountered in two to four individuals per million population per year (4). In the pediatric population, there is a female predominance, and most lesions are extra-abdominal. In patients in their late twenties, there is a female predominance, and lesions are typically of the abdominal wall, whereas in later adult years, there is no sex predisposition and there are proportionally fewer abdominal tumors (5). Tumors are large, and local control can prove difficult, but despite their capacity for local aggression, deep fibromatoses do not metastasize.
Because fibromatoses may be a component of the Gardner syndrome (familial adenomatous polyposis or FAP; 6, 7, 8), it is not surprising that virtually all Gardner syndrome–associated lesions harbor mutations of the APC gene (9, 10, 11, 12, 13, 14, 15). This observation has led to the study of APC mutations in sporadic desmoid tumors. Because one function of the APC gene involves regulation of the cellular level of β-catenin, the interaction between these two molecules has been explored in tandem, and although sporadic desmoid tumors may have APC mutations (16, 17), they are more likely to have β-catenin mutations (18, 19). It appears that the majority of desmoid tumors, both familial and sporadic, have mutations of one of these two genes.
Recently, standard karyotyping analysis of a large series of superficial and deep fibromatoses has revealed clonal chromosomal aberrations in about half of deep fibromatoses and 10% of superficial lesions (20). Loss of 5q, which houses the APC gene, was detected in two of the deep fibromatoses in the latter series but not in superficial lesions. Examples of penile fibromatoses were not included, but cytogenetic aberrations have been described in these (21).
Given the fact that superficial fibromatoses are phenotypically similar to deep fibromatoses in that they are both composed of sweeping fascicles of fibroblasts and myofibroblasts but biologically distinct, we studied β-catenin protein accumulation and β-catenin and APC mutations in a series of these lesions.
MATERIALS AND METHODS
Case Selection
Random cases of superficial fibromatoses with sufficient paraffin-embedded material accessioned between 1994 and 2000 were retrieved from the archives of the Johns Hopkins University Department of Pathology. These years were selected because recent material is less prone to DNA degradation than material from older cases. These included 22 palmar fibromatoses from 21 patients (both of the bilateral lesions were tested in one patient), 5 plantar fibromatoses, 1 infantile digital fibromatosis, and 1 penile fibromatosis. The palmar fibromatoses were from 17 men and 4 women, with a median age of 61 years (range, 23–79 y, mean, 58 y), and bilateral lesions were known to be present in four of the patients. The plantar fibromatoses occurred in three women and two men, at a median age of 47 years (range, 26–60 y, mean, 43 y). The penile fibromatosis was from a 57-year-old man, and the infantile digital fibromatosis was from an 8-year-old boy. None of the patients was known to have the Gardner syndrome. All cases were evaluated by immunohistochemical staining for β-catenin as described below. Five deep fibromatoses were also stained with the β-catenin antibody.
Immunohistochemistry for β-Catenin
Immunoperoxidase stain using diaminobenzidine as the chromogen was performed on the Techmate 1000 automatic staining system (BioTek Solutions, Tucson, AZ). Deparaffinized sections of formalin-fixed tissue were stained with β-catenin antibody (mouse monoclonal antibody, Becton Dickinson Transduction Laboratories, Lexington, KY) at 1:500 dilution after heat-induced antigen retrieval. The percentages of nuclei expressing β-catenin were estimated in each lesion by light microscopy. Normal endothelial cells were used as negative internal controls, and juvenile nasopharyngeal angiofibromas with known β-catenin mutations as positive controls (22).
DNA Extraction
Microdissection of superficial fibromatoses from hematoxylin- and eosin-stained slides for DNA extraction was performed from formalin-fixed, paraffin-embedded specimens. Microdissection was performed using 27-gauge tuberculin needles on un–cover slipped 5-μmsections stained with hematoxylin and eosin under a standard Olympus microscope using the 4× objective. Most of the specimens consisted of lesional tissue alone, but normal skin was dissected in two cases, and skin appendages in four additional cases to serve as nonlesional controls. Genomic DNA was extracted as described elsewhere (23).
Mutation Analysis of the β-Catenin Gene
Direct sequencing of Exon 3 of the β-catenin gene was performed on 26 superficial fibromatosis cases (19 palmar, 5 plantar, 1 penile, and the infantile digital fibromatosis). Genomic DNA from each sample was amplified by polymerase chain reaction (PCR) using the following primer pair: 5′-ATGG- AACCAGACAGAGGGGC-3′ and 5′-GCTACTTGTTC- TGAGTGAAG-3′. These amplified a 200-bp fragment of Exon 3 of the β-catenin gene that encompasses the region for GSK-3β phosphorylation. PCR reaction was performed under standard conditions in a 50-μL volume using PCR Master (Boehringer Mannheim, Mannheim, Germany) and 1 μm of both 5′ and 3′ oligonucleotides with 40 cycles (94° C for 1 minute, 58° C for 1 minute, and 72° C for 2 minutes). PCR products were treated with shrimp alkaline phosphatase and Exonuclease I (Amersham, Buckinghamshire, United Kingdom) before sequencing. Treated PCR products were sequenced directly with SequiTherm EXCEL II DNA Sequencing Kit (Epicentre, Madison, WI) using the internal primers: 5′-AAAGCGGCTGTTAGTCACTFF-3′ and 5′-GACTTGGGAGGTATCCACATCC-3′. Oligonucleotides were end-labeled with (γ-32P)-ATP (NEN DuPont, Boston, MA) using T4 polynucleotide kinase (New England Biolabs, Beverly, MA). These methods have been previously used in our laboratory and are known to readily detect mutations (22, 24, 25).
Mutation Analysis of the APC Gene
Mutation analysis of the APC gene was performed on 20 superficial fibromatosis cases (16 palmar, 3 plantar, 1 penile, and the infantile digital fibromatosis). Four sets of oligonucleotide primers (A1: 5′-CAGACTTATTGTGTAGAAGA-3′ and A2: 5′-CTCC- TGAAGAAAATTCAACA-3′ for Codons 1260 to 1359; B1: 5′-AGGGTTCTAGTTTATCTTCA-3′ and B2: 5′-TCTGCTTGGTGGCATGGTTT-3′ for Codons 1339 to 1436; C1: 5′-GGCATTATAAGCCCCAGTGA-3′ and C2: 5′-AAATGGCTCATCGAGGCTCA-3′ for Codons 1417 to 1516; D1: 5′-ACTCCAGATGGATTTTC- TTG-3′ and D2: 5′-GGCTGGCTTTTTTGCTTTAC-3′ for Codons 1497 to 1596) were used to amplify the mutation cluster region of the APC gene (23). PCR reaction was performed under standard conditions in a 50-μL volume using PCR Master and 1 mM of both 5′ and 3′ oligonucleotides with 40 cycles (94° C for 1 minute, 55° C for 1 minute, and 68° C for 2 minutes for APC-B, C, and D primer pairs and 94° C for 1 minute, 52° C for 1 minute, and 68° C for 2 minutes for APC-A). PCR products were purified using shrimp alkaline phosphatase and exonuclease I. Purified PCR products were sequenced directly with SequiTherm EXCEL II DNA Sequencing Kit using the same primers as for DNA amplification. These methods have been previously used in our laboratory and are known to readily detect mutations (22, 24, 25).
RESULTS
Nuclear Accumulation of β-Catenin
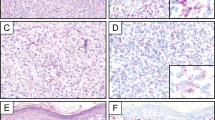
Deep fibromatoses had 60 to 100% nuclear staining in all five cases tested (Fig. 1, A and B). Nuclear accumulation of β-catenin was present in 86% (25/29) of superficial fibromatosis cases ranging from 5 to 100% of nuclei (mean, 13%; median, 10%). With the exception of one case (Case 7), all had a low level of expression. These included 22 palmar fibromatoses from 21 patients (both of the bilateral lesions were tested in one patient), 5 plantar fibromatoses, 1 infantile digital fibromatosis, and 1 penile fibromatosis. The single penile fibromatosis did not express β-catenin, and expression in the infantile digital fibromatosis was focal, although varying degrees of nuclear accumulation were seen in the palmar and plantar lesions (Table 1, Fig. 1, C-F). Endothelial cell nuclei and the nuclei of vascular smooth muscle cells showed cytoplasmic staining but no nuclear β-catenin accumulation, and skin appendages and overlying skin lacked expression when these structures were present.

A and B, deep fibromatosis (desmoid tumor), hematoxylin and eosin (H&E) (A) and β-catenin immunohistochemistry (B). Note that most of the nuclei (estimate: 80%) show β-catenin accumulation, whereas the endothelial cell nuclei and infiltrated skeletal myocyte nuclei are negative. C and D, plantar fibromatosis. These superficial fibromatoses are frequently mitotically active (C, H&E). On the immunohistochemical preparation (D), scattered nuclei (estimate: 10 to 15%) display nuclear β-catenin accumulation, and cytoplasmic staining is focally present. No mutations were identified in either the β-catenin or APC genes. EandF, infantile digital fibromatosis showing the characteristic cytoplasmic inclusions (E, H&E). Scattered nuclei (estimate: 10 to 15% in this field) also show β-catenin accumulation by immunohistochemistry (F), although no mutations were identified in either the β-catenin or APC genes.
Somatic β-Catenin and APC Gene Mutations in Superficial Fibromatoses
No somatic mutations of β-catenin (n = 26) or APC (n = 20) genes in the mutation cluster region were identified in any of the superficial fibromatoses (Table 1).
DISCUSSION
The β-catenin and APC genes are closely interrelated and play a significant role in epithelial neoplasms, best studied in colon cancer (18, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39), but also in deep fibromatoses (19, 40) and sarcomas (41). β-catenin is an intracellular protein that is regulated by the APC protein. The latter complexes with glycogen synthase 3β (GSK-3β) and thus controls degradation of β-catenin. Mutations of the APC gene or the β-catenin gene itself can thus result in the nuclear accumulation of β-catenin protein (42, 43). Because β-catenin is a component of the wnt-signaling pathway controlling cell proliferation, its unregulated accumulation leads ultimately to unchecked cell proliferation and neoplasia. A recently described component of the wnt-signaling pathway is AXIN, and mutations of this group of molecules may be responsible for a subset of colorectal carcinomas through activation of β-catenin signaling (44, 45, 46, 47, 48).
The superficial fibromatoses are quite similar to deep fibromatoses on histomorphologic grounds but differ substantially in their growth characteristics. Because the deep fibromatoses are known to have mutations of either the APC or β-catenin genes, (9, 11, 13, 14, 15, 16, 17, 18, 19, 40, 49, 50, 51, 52) and the status of these genes in superficial fibromatoses was not characterized, we studied the β-catenin status of a group of superficial fibromatoses. We found that despite some degree of nuclear accumulation of β-catenin by immunohistochemical staining, none of the studied superficial fibromatoses had detectable APC or β-catenin mutations. Our results suggest that the APC/β-catenin pathway may play a role in the different growth characteristics of the superficial versus deep fibromatoses and that these two categories of fibromatoses are truly genetically distinct. Our results are also consistent with the recent standard karyotyping series of superficial and deep fibromatoses, which showed greater percentages of anomalies in deep lesions and loss of 5q (the site of the APC gene) only in deep lesions (20).
The cause for the nuclear accumulation of β-catenin in many of our cases by immunohistochemistry is not entirely clear but might be explained by any of several mechanisms. First, superficial fibromatoses could harbor alterations of a gene other than APC or β-catenin in the β-catenin–signaling pathway that was not studied. AXIN genes have recently been shown to be mutated in a subset of hepatocellular carcinomas showing nuclear β-catenin accumulation but lacking β-catenin gene mutations (53). Furthermore, immunohistochemical evidence of β-catenin accumulation does not always correlate well with genetic events (24, 54, 55, 56). Second, because APC is a large gene and only a small fraction of the gene was evaluated, encompassing the mutation cluster region for colorectal adenomas, it is conceivable that other mutations were present in this gene that resulted in β-catenin accumulation. Although the latter possibility cannot be excluded, we have found the techniques used in this series successful in detecting mutations in other organ systems (22, 24, 25, 57), and the same techniques have been applied to the deep fibromatoses, and mutations have been detected in these (9, 10, 11, 12, 13, 14, 15, 17, 40, 49, 51, 52). In contrast to the case with the APC gene, the majority of β-catenin mutations are detectable by sequencing of the GSK-3β phosphorylation region. As such, we believe that these findings reflect true biologic differences between superficial and deep fibromatoses.
Among patients with familial adenomatous polyposis (FAP), intestinal and extraintestinal neoplasms typically arise through biallelic (germline then somatic) inactivation of the APC gene, whereas the corresponding tumors in non-FAP patients occur either through somatic biallelic APC inactivation or somatic mutation of a single β-catenin allele. As the various FAP-associated tumors have been studied, somatic alterations of the APC/β-catenin pathway have been initially detected in familial examples and then subsequently demonstrated in the sporadic counterparts. The first studied tumors were gastrointestinal adenomas (30, 38, 39, 58), followed by desmoid tumors (16, 18, 19), medulloblastomas (59), childhood hepatoblastomas (60, 61), and gastric fundic gland polyps (24, 25). More recently, the same story has unfolded in juvenile nasopharyngeal angiofibromas (22, 62), which occur more frequently in FAP patients than in controls. Sporadic tumors lack APC mutations (63) but are highly likely to have β-catenin mutations (22). It is of interest that superficial fibromatoses are not known to be increased in FAP, even in highly specialized variant disease featuring specific mutations associated with hereditary desmoid tumors (9, 14, 49). It has been estimated that FAP patients in general have an 852-fold increased risk of developing desmoids, typically intraabdominal lesions (64). However, Couture et al. have recently presented a unique French-Canadian kindred harboring a germline mutation of Codons 2643 to 2644 of the APC gene (49). These patients have a penetrance of desmoid tumors approaching 100% and have cutaneous cysts, but few manifest colon polyposis. Noteworthy is that among the fibromatoses found in these specialized carriers, virtually all were axial, and none were superficial. These observations of familial lesions suggest that the pathobiology of deep and superficial fibromatoses differs and that they are truly two divergent and probably biologically unrelated processes. The absence of both β-catenin and APC gene mutations in the studied superficial fibromatoses in contrast to common mutations of these genes in deep fibromatoses reaffirms that the existing classification reflects a biologic distinction that can be confirmed on genetic grounds.
References
Weiss S . Histological typing of soft tissue tumours. Berlin: Springer-Verlag; 1994.
Allen PW . The fibromatoses: a clinicopathologic classification based on 140 cases. Am J Surg Pathol 1977; 1: 255–270.
Iwasaki H, Kikuchi M, Ohtsuki I, Enjoji M, Suenaga N, Mori R . Infantile digital fibromatosis. Identification of actin filaments in cytoplasmic inclusions by heavy meromyosin binding. Cancer 1983; 52: 1653–1661.
Reitamo JJ, Hayry P, Nykyri E, Saxen E . The desmoid tumor. I. Incidence, sex-, age- and anatomical distribution in the Finnish population. Am J Clin Pathol 1982; 77: 665–673.
Hayry P, Reitamo JJ, Totterman S, Hopfner-Hallikainen D, Sivula A . The desmoid tumor. II. Analysis of factors possibly contributing to the etiology and growth behavior. Am J Clin Pathol 1982; 77: 674–680.
Gardner E . A genetic and clinical study of intestinal polyposis, a predisposing factor for carcinoma of the colon and rectum. Am J Hum Genet 1951; 3: 167–176.
Gardner E, Richards R . Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet 1953; 5: 139–147.
Gardner E . Follow-up study of a family group exhibiting dominant inheritance for a syndrome including intestinal polyps, osteomas, fibromas, and epidermal cysts. Am J Hum Genet 1962; 14: 376–390.
Caspari R, Olschwang S, Friedl W, Mandl M, Boisson C, Boker T, et al. Familial adenomatous polyposis: desmoid tumours and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Hum Mol Genet 1995; 4: 337–340.
Davies DR, Armstrong JG, Thakker N, Horner K, Guy SP, Clancy T, et al. Severe Gardner syndrome in families with mutations restricted to a specific region of the APC gene. Am J Hum Genet 1995; 57: 1151–1158.
Eccles DM, van der Luijt R, Breukel C, Bullman H, Bunyan D, Fisher A, et al. Hereditary desmoid disease due to a frameshift mutation at codon 1924 of the APC gene. Am J Hum Genet 1996; 59: 1193–1201.
Enomoto M, Konishi M, Iwama T, Utsunomiya J, Sugihara KI, Miyaki M . The relationship between frequencies of extracolonic manifestations and the position of APC germline mutation in patients with familial adenomatous polyposis. Jpn J Clin Oncol 2000; 30: 82–88.
Halling KC, Lazzaro CR, Honchel R, Bufill JA, Powell SM, Arndt CA, et al. Hereditary desmoid disease in a family with a germline Alu I repeat mutation of the APC gene. Hum Hered 1999; 49: 97–102.
Scott RJ, Froggatt NJ, Trembath RC, Evans DG, Hodgson SV, Maher ER . Familial infiltrative fibromatosis (desmoid tumours) (MIM135290) caused by a recurrent 3′ APC gene mutation. Hum Mol Genet 1996; 5: 1921–1924.
Sen-Gupta S, Van der Luijt RB, Bowles LV, Meera Khan P, Delhanty JD . Somatic mutation of APC gene in desmoid tumour in familial adenomatous polyposis [letter]. Lancet 1993; 342: 552–553.
Alman BA, Li C, Pajerski ME, Diaz-Cano S, Wolfe HJ . Increased beta-catenin protein and somatic APC mutations in sporadic aggressive fibromatoses (desmoid tumors). Am J Pathol 1997; 151: 329–334.
Giarola M, Wells D, Mondini P, Pilotti S, Sala P, Azzarelli A, et al. Mutations of adenomatous polyposis coli (APC) gene are uncommon in sporadic desmoid tumours. Br J Cancer 1998; 78: 582–587.
Miyoshi Y, Iwao K, Nawa G, Yoshikawa H, Ochi T, Nakamura Y . Frequent mutations in the beta-catenin gene in desmoid tumors from patients without familial adenomatous polyposis. Oncol Res 1998; 10: 591–594.
Tejpar S, Nollet F, Li C, Wunder JS, Michils G, dal Cin P, et al. Predominance of beta-catenin mutations and beta-catenin dysregulation in sporadic aggressive fibromatosis (desmoid tumor). Oncogene 1999; 18: 6615–6620.
De Wever I, Dal Cin P, Fletcher CD, Mandahl N, Mertens F, Mitelman F, et al. Cytogenetic, clinical, and morphologic correlations in 78 cases of fibromatosis: a report from the CHAMP Study Group. CHromosomes And Morphology. Mod Pathol 2000; 13: 1080–1085.
Guerneri S, Stioui S, Mantovani F, Austoni E, Simoni G . Multiple clonal chromosome abnormalities in Peyronie's disease. Cancer Genet Cytogenet 1991; 52: 181–185.
Abraham S, Montgomery E, Giardiello F, Wu T . Frequent beta-catenin mutations in juvenile nasopharyngeal angiofibromas. Am J Pathol 2001: 158: 1073–1078.
Moskaluk CA, Kern SE . Microdissection and polymerase chain reaction amplification of genomic DNA from histological tissue sections. Am J Pathol 1997; 150: 1547–1552.
Abraham S, Nobukawa B, Giardiello F, Hamilton S, Wu T . Sporadic fundic gland polyps: common gastric polyps arising through activating mutations in the beta-catenin gene. Am J Pathol 2001: 158: 1005–1010.
Abraham SC, Nobukawa B, Giardiello FM, Hamilton SR, Wu TT . Fundic gland polyps in familial adenomatous polyposis: neoplasms with frequent somatic adenomatous polyposis coli gene alterations. Am J Pathol 2000; 157: 747–754.
Akiyama Y, Nagasaki H, Yagi KO, Nomizu T, Yuasa Y . Beta-catenin and adenomatous polyposis coli (APC) mutations in adenomas from hereditary non-polyposis colorectal cancer patients. Cancer Lett 2000; 157: 185–191.
Herter P, Kuhnen C, Muller KM, Wittinghofer A, Muller O . Intracellular distribution of beta-catenin in colorectal adenomas, carcinomas and Peutz-Jeghers polyps. J Cancer Res Clin Oncol 1999; 125: 297–304.
Hugh TJ, Dillon SA, O'Dowd G, Getty B, Pignatelli M, Poston GJ, et al. Beta-catenin expression in primary and metastatic colorectal carcinoma. Int J Cancer 1999; 82: 504–511.
Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF . Beta-catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci U S A 1997; 94: 10330–10334.
Iwao K, Nakamori S, Kameyama M, Imaoka S, Kinoshita M, Fukui T, et al. Activation of the beta-catenin gene by interstitial deletions involving exon 3 in primary colorectal carcinomas without adenomatous polyposis coli mutations. Cancer Res 1998; 58: 1021–1026.
Kawahara K, Morishita T, Nakamura T, Hamada F, Toyoshima K, Akiyama T . Down-regulation of beta-catenin by the colorectal tumor suppressor APC requires association with Axin and beta-catenin. J Biol Chem 2000; 275: 8369–8374.
Kobayashi M, Honma T, Matsuda Y, Suzuki Y, Narisawa R, Ajioka Y, et al. Nuclear translocation of beta-catenin in colorectal cancer. Br J Cancer 2000; 82: 1689–1693.
Miyaki M, Iijima T, Kimura J, Yasuno M, Mori T, Hayashi Y, et al. Frequent mutation of beta-catenin and APC genes in primary colorectal tumors from patients with hereditary nonpolyposis colorectal cancer. Cancer Res 1999; 59: 4506–4509.
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997; 275: 1787–1790.
Muller O, Nimmrich I, Finke U, Friedl W, Hoffmann I . A beta-catenin mutation in a sporadic colorectal tumor of the RER phenotype and absence of beta-catenin germline mutations in FAP patients. Genes Chromosomes Cancer 1998; 22: 37–41.
Murata M, Iwao K, Miyoshi Y, Nagasawa Y, Yabu M, Himeno S, et al. Activation of the beta-catenin gene by interstitial deletions involving exon 3 as an early event in colorectal tumorigenesis. Cancer Lett 2000; 159: 73–78.
Polakis P, Hart M, Rubinfeld B . Defects in the regulation of beta-catenin in colorectal cancer. Adv Exp Med Biol 1999; 470: 23–32.
Samowitz WS, Powers MD, Spirio LN, Nollet F, van Roy F, Slattery ML . Beta-catenin mutations are more frequent in small colorectal adenomas than in larger adenomas and invasive carcinomas. Cancer Res 1999; 59: 1442–1444.
Sparks AB, Morin PJ, Vogelstein B, Kinzler KW . Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998; 58: 1130–1134.
Li C, Bapat B, Alman BA . Adenomatous polyposis coli gene mutation alters proliferation through its beta-catenin-regulatory function in aggressive fibromatosis (desmoid tumor). Am J Pathol 1998; 153: 709–714.
Kuhnen C, Herter P, Muller O, Muehlberger T, Krause L, Homann H, et al. Beta-catenin in soft tissue sarcomas: expression is related to proliferative activity in high-grade sarcomas. Mod Pathol 2000; 13: 1005–1013.
Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P . Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science 1996; 272: 1023–1026.
Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P . Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci U S A 1995; 92: 3046–3050.
Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, et al. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science 1998; 280: 596–599.
Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A . Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J 1998; 17: 1371–1384.
Liu W, Dong X, Mai M, Seelan RS, Taniguchi K, Krishnadath KK, et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat Genet 2000; 26: 146–147.
Nakamura T, Hamada F, Ishidate T, Anai K, Kawahara K, Toyoshima K, et al. Axin, an inhibitor of the Wnt signalling pathway, interacts with beta-catenin, GSK-3beta and APC and reduces the beta-catenin level. Genes Cells 1998; 3: 395–403.
Spink KE, Polakis P, Weis WI . Structural basis of the Axin-adenomatous polyposis coli interaction. EMBO J 2000; 19: 2270–2279.
Couture J, Mitri A, Lagace R, Smits R, Berk T, Bouchard HL, et al. A germline mutation at the extreme 3′ end of the APC gene results in a severe desmoid phenotype and is associated with overexpression of beta- catenin in the desmoid tumor. Clin Genet 2000; 57: 205–212.
Shitoh K, Konishi F, Iijima T, Ohdaira T, Sakai K, Kanazawa K, et al. A novel case of a sporadic desmoid tumour with mutation of the beta catenin gene. J Clin Pathol 1999; 52: 695–696.
Palmirotta R, Curia MC, Esposito DL, Valanzano R, Messerini L, Ficari F, et al. Novel mutations and inactivation of both alleles of the APC gene in desmoid tumors. Hum Mol Genet 1995; 4: 1979–1981.
Miyaki M, Konishi M, Kikuchi-Yanoshita R, Enomoto M, Tanaka K, Takahashi H, et al. Coexistence of somatic and germ-line mutations of APC gene in desmoid tumors from patients with familial adenomatous polyposis. Cancer Res 1993; 53: 5079–5082.
Clevers H . Axin and hepatocellular carcinomas. Nat Genet 2000; 24: 206–208.
Brabletz T, Herrmann K, Jung A, Faller G, Kirchner T . Expression of nuclear beta-catenin and c-myc is correlated with tumor size but not with proliferative activity of colorectal adenomas. Am J Pathol 2000; 156: 865–870.
Ogawa K, Yamada Y, Kishibe K, Ishizaki K, Tokusashi Y . Beta-catenin mutations are frequent in hepatocellular carcinomas but absent in adenomas induced by diethylnitrosamine in B6C3F1 mice. Cancer Res 1999; 59: 1830–1833.
Palacios J, Gamallo C . Mutations in the beta-catenin gene (CTNNB1) in endometrioid ovarian carcinomas. Cancer Res 1998; 58: 1344–1347.
Choi YW, Heath EI, Heitmiller R, Forastiere AA, Wu TT . Mutations in beta-catenin and APC genes are uncommon in esophageal and esophagogastric junction adenocarcinomas. Mod Pathol 2000; 13: 1055–1059.
Kinzler KW, Vogelstein B . Lessons from hereditary colorectal cancer. Cell 1996; 87: 159–170.
Zurawel RH, Chiappa SA, Allen C, Raffel C . Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Res 1998; 58: 896–899.
Oda H, Imai Y, Nakatsuru Y, Hata J, Ishikawa T . Somatic mutations of the APC gene in sporadic hepatoblastomas. Cancer Res 1996; 56: 3320–3323.
Koch A, Denkhaus D, Albrecht S, Leuschner I, von Schweinitz D, Pietsch T . Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res 1999; 59: 269–273.
Giardiello FM, Hamilton SR, Krush AJ, Offerhaus JA, Booker SV, Petersen GM . Nasopharyngeal angiofibroma in patients with familial adenomatous polyposis. Gastroenterology 1993; 105: 1550–1552.
Guertl B, Beham A, Zechner R, Stammberger H, Hoefler G . Nasopharyngeal angiofibroma: an APC-gene-associated tumor? Hum Pathol 2000; 31: 1411–1413.
Gurbuz AK, Giardiello FM, Petersen GM, Krush AJ, Offerhaus GJ, Booker SV, et al. Desmoid tumours in familial adenomatous polyposis. Gut 1994; 35: 377–381.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Montgomery, E., Lee, JH., Abraham, S. et al. Superficial Fibromatoses are Genetically Distinct from Deep Fibromatoses. Mod Pathol 14, 695–701 (2001). https://doi.org/10.1038/modpathol.3880374
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880374
Keywords
This article is cited by
-
Nuclear TFE3 expression is a diagnostic marker for Desmoid-type fibromatosis
Diagnostic Pathology (2019)
-
T2 signal intensity as an imaging biomarker for patients with superficial Fibromatoses of the hands (Dupuytren’s disease) and feet (Ledderhose disease) undergoing definitive electron beam irradiation
Skeletal Radiology (2018)
