
Abstract
We studied the clinicopathologic and immunohistochemical features of 20 cases of proximal-type epithelioid sarcoma to identify prognostic factors. The 20 patients ranged in age from 13 to 80 years (mean, 40 y); 12 patients were male and 8 were female. The tumors presented as deep soft-tissue or subcutaneous masses on the inguinal region in five, the thigh in four, the vulva in three, the axilla in three, and one each in the flank, chest wall, back, hip and perineum. The tumors ranged from 2 to 16 cm at their greatest diameter (mean: 7.8 cm). Histologically, 12 tumors (60%) were classified as the large-cell subtype, characterized by sheets of large cells with prominent nucleoli resembling poorly differentiated carcinoma, and a frequent rhabdoid phenotype, six (30%) were classified as the conventional subtype, and two (10%) as the angiomatoid subtype. The numbers of tumors exhibiting immunoreactivity for various markers were: vimentin (20 cytokeratin (20 [100%]); epithelial membrane antigen (17 [85%]); CD34 (9 [45%]); CD99 (5 [25%]); muscle markers, either desmin or α-smooth muscle actin (3 [15%]), other markers such as S-100 protein, neurofilament, neuron-specific enolase, synaptophysin and CD56 (12 [60%]); and p53 (16 [80%]). Fourteen lesions (70%) exhibited an MIB-1 index of 30% or more and, by a system of histologic grading using the MIB-1 score, 16 tumors (80%) were classified as high-grade (Grade 3). Thirteen patients (65%) developed local recurrence and 15 (75%) had metastases, primarily to the lymph nodes. At the last follow-up, 13 patients (65%) had died of their disease. A large tumor size and early metastasis were independently associated with a poor outcome. We conclude that proximal-type epithelioid sarcomas are rare, undifferentiated soft-tissue sarcomas of adults, with epithelioid features and a frequent rhabdoid phenotype. These tumors, when arising in proximal locations, have a much worse prognosis than those arising in distal locations.
Similar content being viewed by others
INTRODUCTION
Epithelioid sarcoma, first described by Enzinger in 1970 (1), is a rare soft-tissue sarcoma typically presenting as a subcutaneous or deep dermal mass in distal portions of the extremities of adolescents and young adults. Microscopically, most tumors are characterized by a granuloma-like pattern: nodules of spindled and epithelioid cells circumscribe areas of central hyalinization and necrosis. Fibrous histiocytoma-like and angiomatoid subtypes have also been reported as less common histologic variants (2, 3). Patients often develop multiple local recurrences of long duration, with subsequent metastases in 30 to 50% of cases (4). A more aggressive “proximal” or large-cell type has been described in rare cases to occur as a deep-seated soft-tissue mass at proximal body sites and to show sheets of large cells with prominent nucleoli resembling a poorly differentiated carcinoma and a frequent rhabdoid phenotype (5). Thus, this tumor must be distinguished from other epithelioid lesions, such as extrarenal rhabdoid tumor, synovial sarcoma, angiosarcoma and melanoma, and its prognostic factors have not yet been fully investigated.
We therefore studied 20 cases of proximal or axial epithelioid sarcoma by immunohistochemical analysis to examine the clinicopathologic features that correlate with a poor outcome in patients with these tumors.
MATERIALS AND METHODS
Tumor Samples
We reviewed 20 cases of proximal-type epithelioid sarcoma that were registered in the pathology files of the Clinical Laboratory Division, National Cancer Center Hospital, Tokyo, Japan, and the files of the author (TH). In this study, the diagnostic criteria which were used to select the cases of epithelioid sarcoma arising in proximal locations were the presence of obvious epithelial-like differentiation and malignancy: primarily a solid growth pattern of large epithelioid cells with frequent rhabdoid-appearing cells and a “conventional” granuloma-like pattern, together with immunohistochemical expression of cytokeratin and/or epithelial membrane antigen (EMA). Some of these cases had been diagnosed initially as “rhabdomyosarcoma” (three cases), “synovial sarcoma” (two cases), “granuloma” (one case), “soft-tissue giant cell tumor” (one case), and “undifferentiated carcinoma” (one case; Table 1). There was no history of primary or metastatic carcinoma in our patients. Microscope slides of the primary tumor were reviewed in all cases. Clinical details and follow-up information were obtained by reviewing all medical charts.
Immunohistochemistry and Assessment of Staining
In all 20 cases, representative sections from paraffin blocks were examined by the labeled streptavidin–biotin method, with appropriate use of positive and negative controls throughout. The primary antibodies used in this study and their details are listed in Table 2. Most sections were subjected to heat-induced epitope unmasking in 10 mm citrate buffer, pH 6.0, in an autoclave at 121° C for 20 minutes. All immunostaining was performed on tumor tissue obtained before adjunctive treatment. Staining of more than 10% of the tumor cell nuclei was interpreted as positive for p53.
The Ki-67 (MIB-1 index) was estimated by counting the percentage of positive cell nuclei per 1000 tumor cells in the region of the tumor with the greatest density of staining. The percentages of cells expressing nuclear staining were analyzed as continuous (MIB-1 index) and categorized (MIB-1 score) variables, as shown in Table 3. We used three MIB-1 grading values—those for tumor differentiation, necrosis, and MIB-1 score—as criteria for a histologic grading system (6). A tumor differentiation score of 3 was assigned to epithelioid sarcoma. The separate scores were added together for a combined grade; lesions with a total score of 2 or 3 were classified as Grade 1, those that scored 4 or 5 were Grade 2, and those that scored 6, 7, or 8 were Grade 3. By this MIB-1 grading system, proximal-type epithelioid sarcomas were graded as Grade 2 or 3.
Statistical Analysis
The follow-up period was dated from the time of initial surgery. Survival curves were calculated by the Kaplan–Meier method, and the differences were compared by the log-rank test. Fisher’s exact test was used to examine correlations between the variables. Differences at P < .05 were considered significant.
RESULTS
Clinical Findings
The 20 patients ranged in age from 13 to 80 years (mean, 40 y); 14 patients (70%) were aged 20 to 50 years (Table 1). Twelve patients were male, and eight were female. All patients presented with a palpable mass as the initial symptom. Two patients also had pain. Tumors were located on the inguinal region in five patients, in the thigh in four, the vulva in three, axilla in three, and one each in the flank, chest wall, back, hip, and perineum. Ten tumors involved deep soft tissues and 10 had a subcutaneous or deep dermal location; one of the cases occurring on the vulva (Case 13) appeared as a deeply ulcerated lesion of the skin at the time of recurrence. The tumors ranged from 2 to 16 cm at their greatest diameter (mean: 7.8 cm).
Surgical procedures consisted of wide excision, amputation or disarticulation, with wide margins in eight cases, and marginal or intralesional excision in 12. One patient underwent preoperative radiotherapy and seven were treated postoperatively. Chemotherapy was given preoperatively in four cases, postoperatively in six and both pre- and postoperatively in one. Two patients (Cases 5 and 10) who had metastases at presentation were treated by chemotherapy preoperatively.
Pathologic Findings
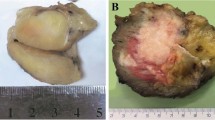
On cut sections, the lesions were ill-defined, multinodular whitish masses, often with hemorrhage and necrosis (Fig. 1).

Recurrent proximal-type epithelioid sarcoma involving the pubis shows ill-defined, multinodular masses in the muscle of the inguinal region (Case 15).
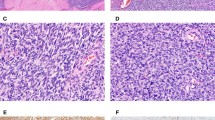
Histologically, 12 tumors (60%) were classified as the large-cell subtype, 6 (30%) as the conventional subtype, and 2 (10%) as the angiomatoid subtype (Table 3). The large-cell epithelioid sarcomas were characterized by sheets of large, rounded polygonal or short spindle cells with enlarged vesicular nuclei, prominent, central single nucleoli and abundant amphophilic cytoplasm (Fig. 2). Rhabdoid cells containing intracytoplasmic, paranuclear hyaline inclusions were abundant in 10 tumors of the large-cell subtype, in three tumors of the conventional subtype, and in one of the angiomatoid subtype. Conventional epithelioid sarcomas displayed nodules of a mixture of atypical spindle and epithelioid cells bordering central areas of hyalinization and necrosis (Fig. 3). Angiomatoid tumors had cystic, blood-filled pseudovascular spaces bordered by epithelioid and spindle cells, similar to those seen in typical cases (Fig. 4).

Proximal-type epithelioid sarcoma of the chest wall (Case 4) shows sheets of large round or polygonal cells with prominent nucleoli (A) and aggregates of rhabdoid cells with prominent cytoplasmic inclusion (B).

Proximal-type epithelioid sarcoma of the back (Case 7) shows a granuloma-like pattern with a mixture of spindled and epithelioid cells surrounding necrotic areas.

Proximal-type epithelioid sarcoma of the perineum (Case 13) shows an angiomatoid appearance characterized by pseudovascular cystic spaces bordered by epithelioid cells.
Vascular invasion was frequently observed, being evident in 11 tumors. Other histologic features present in a minority of cases included scattered, multinucleated osteoclast-like giant cells (three cases), hyalinized collagen (three cases), myxoid areas (three cases), focal small cells (two cases), and central metaplastic bone formation (one case). Hemorrhage and necrotic changes were common findings, and the mitotic count was 20 or more per 10 high-power fields in 15 tumors (75%). Epithelioid sarcoma was given as the initial diagnosis for 12 patients, rhabdomyosarcoma for three, synovial sarcoma for two, and undifferentiated carcinoma, soft-tissue giant cell tumor and granuloma for one each.
Immunohistochemical Findings
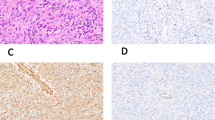
The immunohistochemical results are summarized in Table 3. All 20 cases (100%) showed immunoreactivity for both vimentin and cytokeratin (Fig. 5A). The rhabdoid cytoplasmic inclusions stained variably for vimentin and cytokeratin. EMA was positive in 17 tumors (85%), which displayed moderate to intense cell membrane-based reactivity for EMA. Nine lesions (45%) gave a focal to diffuse membranous positive reaction for CD34 (Fig. 5B). Likewise, focal membranous staining for CD99 (MIC2) was observed in five (25%) lesions. Focal cytoplasmic staining for muscle markers, either desmin or α-smooth muscle actin (SMA), was observed in three (15%) lesions. Twelve cases (60%) were focally positive for other markers, such as S-100 protein, neurofilament, neuron-specific enolase, synaptophysin, and CD56 (N-CAM) (Fig. 5C). Sixteen tumors (80%) yielded positive results for p53 (Fig. 5D). Muscle-specific actin (HHF35), myogenin, CD31, gp100 protein (HMB-45), and chromogranin A were negative in all cases.

Immunohistochemistry of proximal-type epithelioid sarcoma. Many tumor cells show diffuse and strong staining for cytokeratin (A) in Case 3, CD34 (B) in Case 13, neurofilament (C) in Case 14, and p53 (D) in Case 12.
MIB-1 immunoreactivity was present in all 20 cases, and the MIB-1 index ranged from 5% to 90% (mean, 50%). Fourteen lesions (70%) exhibited an MIB-1 index of 30% or more and, by our histologic grading system using the MIB-1 score, 16 tumors (80%) were classified as Grade 3, and 4 (20%) as Grade 2.
Follow-up and Prognostic Analysis
Follow-up information on all 20 patients was available, and covered periods ranging from 2 months to 19 years (mean, 4 y and 5 mo). Thirteen patients (65%) developed local recurrence; the period between primary excision and the first local recurrence ranged from 1 month to 3 years and 8 months (mean, 10 mo). Most patients had multiple recurrences; notably, one patient (case 2) experienced 11 recurrences over 10 years. Fifteen patients (75%) developed metastases, primarily to the lymph node (12 cases), lung (seven), bone (four), and skin (two). The time interval from surgery to the first metastasis ranged from within 1 month to 12 years (mean, 2 y and 4 mo). At the last follow-up, 13 patients (65%) had died of the disease, and one had died of other causes. Six patients (30%) were alive with no evidence of disease.
Survival of patients whose neoplasms measured 7.8 cm or more (9 cases) was significantly shorter than for those with smaller tumors (P = .0027). Survival of patients whose first metastasis occurred within 28 months (10 cases) was also significantly shorter than for those with more than 29 months before metastasis or those without metastasis (P = .0026). There was no association between tumor size and metastasis. Patients whose tumors were excised marginally survived longer than those who had wide excision (P = .0091), but surgical margin was inversely correlated with tumor size (P = .0045); marginal excision, frequently followed by chemotherapy or radiation therapy, was employed mostly for the small tumors. Kaplan–Meier survival curves were plotted for all 20 patients, according to tumor size and metastasis (Fig. 6). No other categorized variables, including age (≥40 or <40 years), sex, depth, adjuvant therapy, recurrence, histologic subtype (large-cell or others), presence of rhabdoid cells, vascular invasion, tumor necrosis, mitotic count (≥20 or <20 per 10HPF), p53 expression, MIB-1 index (≥30 or <30%) and MIB-1 grade correlated with overall survival.

Survival curves of proximal-type epithelioid sarcoma by tumor size (A) and metastasis (B).
DISCUSSION
The clinical, morphological and immunohistochemical features of the 20 tumors described here are in accordance with those of proximally located epithelioid sarcomas (5). The tumors in this study arose predominantly in the deep soft-tissue or subcutis of young adults or middle-aged individuals and had a significant male predominance. Large tumors with a maximum diameter of more than 10 cm were noted at the thigh, hip and flank in this series. Histologically distinctive features were prominence of the epithelioid cell component, a sheet-like growth pattern of large cells with vesicular nuclei and prominent nucleoli, and a frequent rhabdoid appearance. As described previously (7), however, epithelioid sarcomas with a typical granuloma-like pattern and other histologic subtypes were also present at proximal locations in our series.
Immunohistochemically, irrespective of the tumor types, the proximal-type epithelioid sarcomas of our series were clearly epithelial-like, with cytokeratin and EMA commonly present, and CD34 in about half of the cases. These tumors showed cell membrane-based reactivity for EMA. It should be noted that many soft-tissue sarcomas, except for synovial sarcoma and epithelioid sarcoma, demonstrate nonspecific cytoplasmic EMA-reactivity (8). A few tumors were positive for desmin and SMA. Focal immunostaining for CD99 and other markers was much more common in the large-cell variant than in other forms. Such a polyphenotypic profile has been described in conventional and proximal, or large-cell type, epithelioid sarcomas (5, 7) as well as in extrarenal rhabdoid tumors (9, 10).
The pathognomonic feature of the latter tumor is the presence of rhabdoid cells with intracytoplasmic inclusions, which prove ultrastructurally to be whorls of intermediate filaments. This tumor type has been documented in a variety of sarcomas and also in carcinomas and melanomas (11). Its distinction from proximal-type epithelioid sarcoma may be difficult at times. Extrarenal rhabdoid tumor, like the renal counterpart, is a highly aggressive and lethal neoplasm occurring in younger children and has inactivating mutations or deletions of both alleles of the tumor suppressor gene, hSNF5/INI1 on chromosome 22q11.2 (12). Considering that this chromosomal aberration involving chromosome 22 may be an epiphenomenon rather than a primary pathogenetic event, it is likely that proximal-type epithelioid sarcoma may represent a form of “composite” extrarenal rhabdoid tumor (11).
Given the varied morphologic features and immunophenotypic heterogeneity of the present cases, proximal-type epithelioid sarcomas can potentially be mistaken for other epithelioid malignancy. Epithelioid malignant peripheral nerve sheath tumors (MPNSTs) might be positive for cytokeratin and EMA occasionally, but about 80% of cases of MPNST show diffuse and strong reactivity for S-100 protein (13). Similarly, malignant melanomas are typically strongly immunoreactive for S-100 protein and gp100 protein (HMB-45).
Epithelioid morphology can occasionally be seen in both rhabdomyosarcomas and leiomyosarcomas. It has also been reported that approximately 30% of leiomyosarcomas are immunoreactive for cytokeratin and EMA (14). However, epithelioid sarcomas are easily distinguished from leiomyosarcomas by their lacking a representative area of a leiomyosarcoma, showing fascicles of elongated tumor cells with blunt-ended “cigar-shaped” atypical nuclei, and greater frequency of negativity for desmin and SMA. Evidence of rhabdomyoblastic differentiation appreciated by positivity for HHF35 and myogenin (15) was absent in epithelioid sarcomas.
Angiomatoid epithelioid sarcomas with a pseudovascular pattern in our series may be easily confused with epithelioid angiosarcoma, which may be positive for cytokeratin and CD34 (16, 17). Also, many epithelioid angiosarcomas have a diffuse sheet-like growth pattern, and a vasoformative architecture is often not present. They have positive immunoreactivity for a sensitive and specific marker for endothelial differentiation, CD31 (18). In addition, epithelioid angiosarcomas have few or no immunoreactivity for EMA.
Synovial sarcomas display an epithelial immunophenotype with positivity for both cytokeratin and EMA, and also have occasional reactivity for neural markers (19), as found in our series. However, synovial sarcomas usually show, at least focally, a biphasic pattern allowing correct diagnosis, and are consistently negative for CD34 (18). High-grade large epithelioid cells and a rhabdoid phenotype observed in extraskeletal myxoid chondrosarcomas may cause confusion with epithelioid sarcoma (20). Neuroendocrine differentiation was also reported in this tumor type (21). The presence of the lobular architecture and typical appearance of cords and strands of eosinophilic chondroblasts embedded in a myxoid matrix readily distinguish extraskeletal myxoid chondrosarcoma from epithelioid sarcoma.
The distinction between proximal-type epithelioid sarcoma and undifferentiated carcinoma is probably the most difficult consideration. The occurrence of tumors in the subcutis or deep soft tissues without any connection with the overlying epidermis or cutaneous adnexa, the absence of histologic features of squamous or glandular differentiation, and presence of CD34 reactivity in about half of the cases favor the diagnosis of epithelioid sarcoma over undifferentiated carcinoma. The latter are negative for CD34 in most cases (18).
Most tumors had high MIB-1 indexes and were assessed as high grade by our histologic grading using the MIB-1 staining. Our study also revealed that p53 nuclear accumulation was a conspicuous feature in 80% of tumors. We employed an anti-p53 rabbit polyclonal antibody (Rsp53, Nichirei Co., Tokyo, Japan) to increase sensitivity, but there was no difference of the frequency of positivity between polyclonal and monoclonal DO7 antibodies (data not shown). The increased p53 overexpression is probably associated with the higher histologic grade of proximal-type epithelioid sarcomas because p53 did correlate with grade in a previous study of 95 adult soft-tissue sarcomas (6).
Our study clearly showed that proximally or axially located epithelioid sarcomas behaved more aggressively than conventional distally located tumors; 65% of the patients in our series have died of disease. In previous series of conventional epithelioid sarcomas (4, 22, 23), 27–46% of the patients have died of disease. The rate of tumor-related deaths is lower than the rate of metastasis (75%), which indicates the indolent yet persistent nature of this tumor. The adverse prognostic factor was larger tumor size as documented by others (4, 22, 23). Also, early metastasis independently correlated with clinical aggressiveness. Other parameters, including patient age and sex, tumor depth, treatment-related factors, recurrence, histologic subtype, rhabdoid cells, vascular invasion, tumor necrosis, mitotic count, p53 expression, MIB-1 index and MIB-1 grade, had no significant relationship with patient survival. However, the case numbers in this series are relatively small, and further study is necessary to evaluate prognostic impact of these parameters in a fairly large number of proximal-type epithelioid sarcomas.
CONCLUSION
We conclude that proximal-type epithelioid sarcomas are rare, undifferentiated soft-tissue sarcomas of adults, with epithelioid features and a frequent rhabdoid phenotype. These tumors, when arising in proximal locations, have a much worse prognosis than those arising in distal locations.
References
Enzinger FM . Epithelioid sarcoma: a sarcoma simulating a granuloma or carcinoma. Cancer 1970; 26: 1029–1041.
von Hochstetter AR, Meyer VE, Grant JW, Honegger HP, Schreiber A . Epithelioid sarcoma mimicking angiosarcoma: the value of immunohistochemistry in the differential diagnosis. Virchows Arch A Pathol Anat Histopathol 1991; 418: 271–278.
Mirra JM, Kessler S, Bhuta S, Eckardt J . The fibroma-like variant of epithelioid sarcoma: a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer 1992; 69: 1382–1395.
Chase DR, Enzinger FM . Epithelioid sarcoma. Diagnosis, prognostic indicators, and treatment. Am J Surg Pathol 1985; 9: 241–263.
Guillou L, Wadden C, Coindre JM, Krausz T, Fletcher CDM . “Proximal-type” epithelioid sarcoma: a distinctive aggressive neoplasm showing rhabdoid features. Clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol 1997; 21: 130–146.
Hasegawa T, Yokoyama R, Lee YH, Shimoda T, Beppu Y, Hirohashi S . Prognostic relevance of a histological grading system using MIB-1 for adult soft-tissue sarcoma. Oncology 2000; 58: 66–74.
Miettinen M, Fanburg-Smith JC, Virolainen M, Shmookler BM, Fetsch JF . Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol 1999; 30: 934–942.
Swanson PE, Scheithauer BW, Manivel JC, Wick MR . Epithelial membrane antigen reactivity in mesenchymal neoplasms: an immunohistochemical study of 306 soft tissue sarcomas. Surg Pathol 1989; 2: 313–322.
Parham DM, Weeks DA, Beckwith JB . The clinicopathologic spectrum of putative extrarenal rhabdoid tumors. An analysis of 42 cases studied with immunohistochemistry or electron microscopy. Am J Surg Pathol 1994; 18: 1010–1029.
Fanburg-Smith JC, Hengge M, Hengge UR, Smith JSC, Miettinen M . Extrarenal rhabdoid tumors of soft tissue: a clinicopathologic and immunohistochemical study of 18 cases. Ann Diagn Pathol 1998; 2: 351–362.
Wick MR, Ritter JH, Dehner LP . Malignant rhabdoid tumors: a clinicopathologic review and conceptual discussion. Semin Diagn Pathol 1995; 12: 233–248.
Ogino S, Ro JY, Redlinek RW . Malignant rhabdoid tumor: a phenotype? an entity?—a controversy revisited. Adv Anat Pathol 2000; 7: 181–190.
Scheithauer BW, Woodruff JM, Erlandson RA . Primary malignant tumors of peripheral nerve. In: Scheithauer BW, Woodruff JM, Erlandson RA, editors. Tumors of the peripheral nervous system. Atlas of tumor pathology. Fascicle 24. Washington, DC: Armed Forces Institute of Pathology; 1999. p 303–372.
Iwata J, Fletcher CDM . Immunohistochemical detection of cytokeratin and epithelial membrane antigen in leiomyosarcoma: a systematic study of 100 cases. Pathol Int 2000; 50: 7–14.
Kumar S, Perlman E, Harris CA, Raffeld M, Tsokos M . Myogenin is a specific marker for rhabdomyosarcoma: an immunohistochemical study in paraffin-embedded tissues. Mod Pathol 2000; 13: 988–993.
Fletcher CDM, Beham A, Bekir S, Clarke AMT, Marley NJE . Epithelioid angiosarcoma of deep soft tissue: a distinct tumor readily mistaken for an epithelial neoplasm. Am J Surg Pathol 1991; 15: 915–924.
Meis-Kindblom JM, Kindblom LG . Angiosarcoma of soft tissue: a study of 80 cases. Am J Surg Pathol 1998; 22: 683–697.
Miettinen M, Lindenmayeer E, Chaubal A . Endothelial cell markers CD31, CD34, and BNH9 antibody to H- and Y-antigens-evaluation of their specificity and sensitivity in the diagnosis of vascular tumors and comparison with von Willebrand factor. Mod Pathol 1994; 7: 82–90.
Folpe AL, Schmidt RA, Chapman D, Gown AM . Poorly differentiated synovial sarcoma: immunohistochemical distinction from primitive neuroectodermal tumors and high-grade malignant peripheral nerve sheath tumors. Am J Surg Pathol 1998; 22: 647–682.
Lucas DR, Fletcher CDM, Adsay NV, Zalupski MM . High-grade extraskeletal myxoid chondrosarcoma: a high-grade epithelioid malignancy. Histopathology 1999; 35: 201–208.
Harris M, Coyne J, Tariq M, Eyden BP, Atkinson M, Freemont AJ, et al. Extraskeletal myxoid chondrosarcoma with neuroendocrine differentiation: a pathologic, cytogenetic, and molecular study of a case with a novel translocation t(9;17)(q22;q11.2). Am J Surg Pathol 2000; 24: 1020–1026.
Evnas HL, Baer SC . Epithelioid sarcoma: a clinicopathologic and prognostic study of 26 cases. Semin Diagn Pathol 1993; 10: 286–291.
Halling AC, Wollan PC, Pritchard DJ, Vlasak R, Nascimento AG . Epithelioid sarcoma: a clinicopathologic review of 55 cases. Mayo Clin Proc 1996; 71: 636–642.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hasegawa, T., Matsuno, Y., Shimoda, T. et al. Proximal-Type Epithelioid Sarcoma: A Clinicopathologic Study of 20 Cases. Mod Pathol 14, 655–663 (2001). https://doi.org/10.1038/modpathol.3880368
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880368
Keywords
This article is cited by
-
Primary adult sellar SMARCB1/INI1-deficient tumor represents a subtype of atypical teratoid/rhabdoid tumor
Modern Pathology (2022)
-
Mesenchymal/non-epithelial mimickers of neuroendocrine neoplasms with a focus on fusion gene-associated and SWI/SNF-deficient tumors
Virchows Archiv (2021)
-
Proximal-Type Epithelioid Sarcoma in Skull Base: a Pathological Diagnosis Challenge with Other Intracranial Tumors
Pathology & Oncology Research (2019)
-
Epithelioid sarcoma in the chest wall: a case report and literature review
Surgical Case Reports (2018)
-
SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus): a clinicopathologic entity distinct from undifferentiated carcinoma
Modern Pathology (2018)
