
Abstract
Anaplastic large cell lymphoma (ALCL) is a paradigm for the process used to define new disease entities, and provides a model that is applicable to all areas of pathology. ALCL was first recognized based on characteristic histologic features (sinusoidal invasion) and a distinctive immunophenotype (CD30+). However, neither sinusoidal invasion nor CD30-positivity proved to be entirely specific. Subsequently, a characteristic cytogenetic abnormality was identified, the t(2;5), that led to identification of the genes involved in the translocation (NPM/ALK) and insights into the pathogenesis. Generation of monoclonal antibodies to the aberrantly expressed anaplastic large cell lymphoma kinase (ALK) such as ALK-1 can be used diagnostically, and have led to improved definition of the diagnostic entity with important clinical and prognostic implications. These studies also have clarified the relationship of ALCL to Hodgkin's disease, another lymphoid malignancy associated with CD30 expression. We have learned that the ultimate histologic spectrum of ALCL is both narrower and broader than originally believed. The small cell and lymphohistiocytic variants of ALCL are ALK-positive, and are an accepted part of the disease entity, although the neoplastic cells may appear neither large nor anaplastic. Conversely, most cases of Hodgkin's-like ALCL have proved to be more closely related to true Hodgkin's disease, and are unrelated to ALCL.
Similar content being viewed by others

INTRODUCTION
Anaplastic large cell lymphoma (ALCL) is a paradigm for the process used to define new disease entities, and provides a model that is applicable to all areas of pathology. ALCL was first recognized based on characteristic histologic features (sinusoidal invasion) and a distinctive immunophenotype (CD30+) (Fig. 1) (1). However, neither sinusoidal invasion nor CD30-positivity proved to be entirely specific. Subsequently, a characteristic cytogenetic abnormality was identified, the t(2;5), that led to identification of the genes involved in the most common translocation of ALCL (NPM/ALK) and insights into the pathogenesis (2, 3, 4). Generation of monoclonal antibodies to the aberrantly expressed ALCL tyrosine kinase or ALK (ALK-1, ALKc) can be used diagnostically, and have led to improved definition of the diagnostic entity with important clinical and prognostic implications (5). Utilizing these biologic tools, the ultimate histologic spectrum of ALCL is both broader and narrower than originally believed. It is likely that our ultimate definition of this disease will be restricted to “T-cell” lymphomas with aberrant expression of ALK.

This schematic diagram tracks the evolution of ALCL to our current understanding. The initial descriptions of ALCL were based on the expression of the Ki-1 antigen and sinusoidal infiltration by tumor cells. ALCL was postulated to be related to HD, PTCL, and MH. Further immunophenotypic studies identified ALCL as a unique type of T-cell lymphoma. Investigation of the molecular pathogenesis of ALCL led to the development of new diagnostic tools. These diagnostic tools have helped to resolve the borderlands of the entity, and led to improved morphologic criteria for diagnosis, with the recognition of hallmark cells. HD, Hodgkin's disease; PTCL, peripheral T-cell lymphoma; MH, malignant histiocytosis.
HISTORICAL BACKGROUND
The historical background of ALCL begins in 1982 when the monoclonal antibody Ki-1 was raised against a Hodgkin's disease (HD) cell line (Table 1) (6). Ki-1 was subsequently shown to stain the malignant cells of HD in tissue sections, as well as a smaller lymphoid cell found in the parafollicular regions of normal lymph nodes (7). It was postulated that this CD30+ cell might represent a precursor lymphoid cell for the malignant cells of HD. The Ki-1 antigen, subsequently assigned to the CD30 cluster, was identified as an activation antigen, which could be expressed on T-cells, B-cells, and even activated histiocytes (8, 9, 10). It is part of the nerve growth factor/tumor necrosis factor superfamily, and the gene encoding CD30 is found on chromosome 1p36 (11). The CD30 antigen is found in elevated levels in the serum of patients with HD and other CD30-expressing malignancies (12, 13).
In 1985, Stein et al. found that the Ki-1 (CD30) antigen was highly expressed by a group of 45 large cell lymphomas that shared a number of histologic characteristics (1). These tumors exhibited a morphology suggestive of “malignant histiocytes,” exhibiting prominent sinusoidal invasion. Ki-1 also stained cases of lymphomatoid papulosis, 28% of peripheral T-cell lymphomas, and a small percentage of large B-cell lymphomas. It was proposed that the anaplastic CD30-positive tumors represented a distinct clinicopathologic entity, subsequently included in the revised Kiel classification in 1988 as “large cell anaplastic lymphoma” (14, 15). In 1986, Kadin et al. described a similar lesion involving the lymph nodes and skin in six pediatric patients under the designation “childhood Ki-1 lymphoma” (16). Ultimately, the term “anaplastic large cell lymphoma” has become the preferred designation (17).
There has been some confusion regarding the significance of the designation “anaplastic” for this neoplasm. The term “anaplastic” referred to the fact that the neoplastic cells grew in a manner unlike normal T or B-lymphocytes, and did not resemble any normal lymphoid cell (1). The cytology ranged from an immunoblastic appearance, to one characterized by more bizarre cells with abundant basophilic cytoplasm. Although Stein et al. and others initially postulated that the neoplastic cells might be related to Reed-Sternberg cells (18), a biologic relationship to HD eventually was disproved (19, 20, 21). Moreover, multinucleated cells resembling Reed-Sternberg cells are uncommon in ALCL.
Although the original series of Stein et al. included tumors expressing either T-cell or B-cell associated antigens, the final definition of ALCL as proposed by the International Lymphoma Study Group in the Revised European-American Lymphoma classification was restricted to lymphomas with a T-cell or null cell phenotype (17). Based on extensive biologic and clinical studies, B-cell lymphomas expressing the CD30 antigen were felt to be unrelated. As noted above, the CD30 antigen is an activation antigen, which can be expressed on either T-cells or B-cells. B-cell derived lymphoid malignancies frequently expressing CD30 include HD, Epstein-Barr virus (EBV) positive lymphomas, primary effusion lymphomas, and B-cell tumors with plasmacytoid differentiation (22). In addition, there are some large B-cell lymphomas with “anaplastic cytology” and CD30 expression that are considered a morphologic variant of large B-cell lymphoma (17, 23, 24).
The pathogenesis of ALCL began to be unraveled when in 1989 the t(2;5)(p23q35) formerly associated with cases of “malignant histiocytosis” was found in ALCL (2, 3). Morris and colleagues cloned the genes involved in the translocation in 1994, identifying the gene on chromosome 2 as a newly described tyrosine kinase, named the anaplastic large cell lymphoma kinase (ALK) (4). The partner in this translocation was the nucleophosmin (NPM) gene, although subsequently it was shown that ALK could have other translocation partners. To aid in the diagnosis of ALCL, polyclonal and monoclonal antibodies to the ALK kinase were raised (5, 25). In particular the ALK-1 monoclonal antibody raised against a formalin resistant epitope of ALK has proven extremely useful for clinical studies and routine diagnosis (26, 27).
HISTOLOGIC FEATURES
In the common type of ALCL, the neoplastic cells generally have abundant basophilic cytoplasm that appears gray-blue in hematoxylin and eosin stained sections (Fig. 2). Usually a prominent Golgi zone is evident, staining as a clear or more eosinophilic zone in tissue sections. The nucleus often is lobulated (horseshoe or kidney shaped), with the nuclear lobes surrounding the Golgi region. Delsol has referred to cells with these cytologic features as “hallmark” cells, because they constitute a hallmark of ALCL (26). Although they are typically large cells, smaller cells with similar cytologic features also may be seen (28). With practice, recognition of these “hallmark” cells can greatly aid in accurate diagnosis. Depending on the plane of section, some cells may appear to contain cytoplasmic inclusions, but these are not true inclusions but invaginations of the nuclear membrane. These cells have been referred to as “doughnut” cells (29). Although the horseshoe shaped cell is the hallmark cell, the nuclear shape can vary. In many cases the nuclei are round to oval in shape, and relatively monomorphic (19, 27, 30). The nuclear chromatin is finely clumped, dispersed in a clear parachromatin. The cells usually contain multiple small basophilic nucleoli. Prominent inclusion-like nucleoli are relatively uncommon, aiding in the differential diagnosis with HD (31).

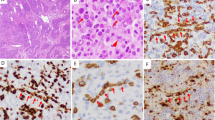
Histologic spectrum of ALCL. A, Cells are large with abundant amphophilic cytoplasm and distinct cytoplasmic borders. A cohesive growth pattern is present. The cells are relatively monomorphic, although some indented cells resembling hallmark cells are seen. B, Hallmark cells have lobulated nuclei with prominent Golgi zones. C, In the lymphohistiocytic variant the neoplastic cells are overshadowed by histiocytes with eosinophilic cytoplasm. D, A hallmark cell is seen in a background of histiocytes, same case as Figure C. E, In the small cells variant neoplastic cells have small irregular nuclei and pale cytoplasm. Neoplastic cells may be contrasted with normal lymphocytes in reactive paracortex at lower left. F, Perivascular rosetting by tumor cells is a common feature. G, Some cases of ALCL may have a sarcomatoid appearance with edematous stroma. H, At higher power neoplastic cells are associated with fibroblasts containing plump nuclei.
A notable diagnostic feature is the propensity of ALCL cells to invade lymphoid sinuses, although in many cases complete architectural effacement can be seen (30). Another helpful architectural feature is the tendency of malignant cells to form rosettes around blood vessels (26, 28). In the small cell and lymphohistiocytic variants of ALCL, the larger cells usually will be concentrated around blood cells, a distribution highlighted by staining for CD30 or ALK. The tumor cells generally are cohesive. Variable architectural features include nodal and capsular fibrosis. However, well-formed fibrous bands are generally absent, in contrast to HD.
An inflammatory background is variable, and is usually more evident in histologic variants containing smaller lymphoid cells. Histiocytes with round nuclei and abundant eosinophilic cytoplasm may be abundant and may mask the neoplastic cells (32, 33). The histiocytes are uniform in appearance; multinucleated giant cells or epithelioid histiocytes forming granulomas are absent. Plasma cells may be numerous, but eosinophils are infrequent. In most cases polymorphonuclear leukocytes are absent, although rare cases containing abundant neutrophils or eosinophils have been described (34, 35).
The common type of ALCL is not surprisingly the most common, constituting 75% or more of the cases (26). However, other histologic variants may be seen, either alone or in sequence with the common type of ALCL. The small cell and lymphohistiocytic variants are the most common, and are particularly important because they may be misdiagnosed as either atypical inflammatory lesions or malignant lymphomas of other types (28, 32). The small cell and lymphohistiocytic variants are closely related, and cases containing a mixture of these two patterns can be seen. These are the only variants specified in the World Health Organization classification of hematopoietic and lymphoid neoplasms (36).
In the small cell variant, the predominant neoplastic cell is small with clear cytoplasm (28). The nucleus has clumped chromatin, and frequent nuclear irregularities, resembling the horseshoe shaped nuclei of classical hallmark cells. The cells have distinct cytoplasmic membranes. Although in routine tissue sections these cells may be mistaken for histiocytes, they will be negative for CD68 and lysozyme by immunohistochemical studies. A diagnosis of peripheral T-cell lymphoma, unspecified may be suspected. However, the neoplastic cells are usually negative for CD3 and often negative for CD45RO; staining for CD30 or ALK will aid in accurate diagnosis.
The lymphohistiocytic variant contains similar small cells admixed with abundant pale histiocytes (32). The histiocytes may be so abundant as to mask a small population of tumor cells in the background. Plasma cells are often present; however, neutrophils and eosinophils are sparse to absent. In the lymphohistiocytic variant, the neoplastic cells include hallmark cells, as well as small neoplastic cells, resembling those of the small cell variant. Therefore, the small cell and lymphohistiocytic variants appear closely related. In both variants, some classical hallmark cells are invariably found, often forming perivascular rosettes around blood vessels.
The sarcomatoid variant is relatively rare, and may simulate a soft tissue sarcoma (37). Plump and proliferating fibroblasts are numerous. The stroma is usually edematous, and may contain admixed inflammatory cells. Neoplastic cells are distributed in this background, again sometimes forming perivascular rosettes. This variant must be distinguished from inflammatory myofibroblastic tumors in children, which recently have been reported to express ALK as a consequence of translocations involving the tropomyosin gene and the ALK tyrosine kinase (38, 39). Immunohistochemical studies are critical in this differential diagnosis. The neoplastic cells in ALCL express both CD30 and ALK. In inflammatory myofibroblastic tumors carrying the ALK translocation, the spindle cells will be ALK-positive but CD30-negative.
So-called “Hodgkin's-like ALCL” is a variant that also has undergone reassessment. Although originally proposed as a variant of ALCL resembling HD (40, 41), it is now felt that these cases can be resolved as either Hodgkin's lymphoma or ALCL with the use of a battery of immunohistochemical and, if needed, molecular techniques. Most cases to which this term was previously applied are felt to be examples of histologically aggressive Hodgkin's lymphoma, either nodular sclerosis type II, or lymphocytic depletion. These cases contain sheets of Hodgkin's cells and lacunar cells palisaded around areas of necrosis. The neoplastic cells, as expected, are CD30 positive, but CD15 expression is more variable. For these reasons, the Hodgkin's-like variant has not been retained in the World Health Organization classification of lymphoid neoplasms (42). However, rare cases of ALCL may contain occasional cells resembling Reed-Sternberg cells.
The use of immunohistochemistry for CD30 and ALK has both broadened and narrowed our definition of ALCL. It has proved that the common, small cell and lymphohistiocytic variants are part of a single clinicopathologic entity (26, 31, 33). These histologic patterns may be seen in the same patient at different points in time. The different histologic variants do not appear to have distinct prognostic significance, and thus should not be considered “histologic grades” or “prognostic variants.” Less common histologic patterns of ALCL including those containing giant cells, a sarcomatoid growth pattern, or Hodgkin's-like cells are less frequently ALK-positive, and thus many of these cases are probably not bona fide examples of ALCL.
CLINICAL FEATURES
ALCL is most common in the pediatric, adolescent, and young adult age groups (26, 43, 44, 45). In some series a bi-modal age distribution is seen (19, 30, 46). However, cases of ALCL in the elderly usually are t(2;5) or ALK negative, and probably constitute an unrelated clinicopathologic entity. There is a marked male predominance, up to 6:1 in some series (43). ALCL usually presents with lymph node enlargement. Frequent extranodal sites of disease include skin, bone, and soft tissue (47). Soft tissue masses may sometimes be mistaken for soft tissue sarcomas. Involvement of other extranodal sites is less commonly seen. ALCL has an aggressive natural history, but an excellent response to combination chemotherapy containing doxorubicin. It is one of the most curable of the aggressive non-Hodgkin's lymphomas (48). Notably, it has a much better prognosis than any other type of T-cell lymphoma.
Although skin involvement may be seen at diagnosis or relapse in classical systemic ALCL, primary cutaneous ALCL is an unrelated disease entity with a different pathogenesis and different clinical behavior (42, 49, 50, 51, 52, 53, 54). Primary cutaneous ALCL is part of the spectrum of CD30+ T-cell lymproliferative disease of the skin, including both lymphomatoid papulosis and borderline lesions (55). A detailed discussion of primary cutaneous ALCL is beyond the scope of this review. However, it should be noted that primary cutaneous ALCL is ALK-negative, and often negative for epithelial membrane antigen (EMA) as well.
Systemic symptoms are common in ALCL. The international prognostic index appears to be of some value in predicting outcome, although less so than in other variants of lymphoma (43, 44, 48). The most important prognostic indicator is ALK positivity, which has been associated with a favorable prognosis in series from the United States, Europe, and Japan (44, 56, 57). Therefore, ALK staining is an important tool, both for accurate diagnosis and as a guide to prognosis.
Bone marrow involvement is relatively common in ALCL, but may be easily missed in routine trephine sections or aspirate smears (58). The neoplastic cells usually infiltrate as single cells, and are most readily identified with stains for CD30, EMA, or ALK. As the smaller tumor cells may be CD30-negative, staining for ALK with either ALK-1 or ALKc is preferable. In rare cases peripheral blood involvement may also be seen. Because bone marrow involvement has been an adverse risk factor, immunohistochemical stains of staging bone marrow biopsies should be performed.
IMMUNOPHENOTYPE
Immunophenotypic studies are extremely useful in the accurate diagnosis of ALCL, and one could argue that the diagnosis should not be made without immunophenotypic support (Table 2) (48). The hallmark of ALCL is expression of CD30, but of course CD30 expression is not specific for ALCL and can be seen in activated lymphoid cells, other T and B cell lymphomas, HD, and even germ cell neoplasms (Fig. 3) (1, 8, 10, 59, 60). For these reasons, the term Ki-1 lymphoma is not favored, as the use of this term implies specificity of the CD30 antigen for this entity (61). EMA positivity is seen in the majority of cases, and correlates roughly with the expression of ALK (62). Because of the prominent sinusoidal localization, keratin stains rather than EMA should be used if the differential diagnosis includes metastatic carcinoma. Staining for the ALK kinase is one of the most specific diagnostic tools, and is positive in more than 80% of cases diagnosed as ALCL by other stringent criteria (19, 26).

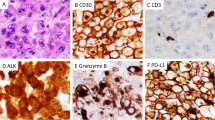
Immunohistochemical findings in ALCL. A, CD30 highlights tumor cells surrounding blood vessel (same case as shown in Figure 1, G and H). B, EMA stains tumor cells located in sinusoidal space. C, In this case with a classic t(2;5) ALK stains both cytoplasm and nuclei. Nucleoli are stained as well. D, ALCL cells usually manifest a cytotoxic phenotype, demonstrated here with stain for TIA-1.
By molecular analysis ALCL can usually be shown to be of T-cell origin (63, 64, 65). However, the cells exhibit an aberrant phenotype with loss of many of the T-cell associated antigens (19). Both CD3 and CD45RO, the most widely used pan T-cell markers are negative in greater than 50% of cases (62). CD5 and CD7 are often negative as well. CD43 is positive in a higher proportion, but this antigen lacks lineage specificity. CD2 and CD4 are positive in the majority of cases, and antibodies to both of these antigens are now available and reliable. CD8 is usually negative, but rare CD8-positive cases exist. ALCL cells, despite the usual CD4+/CD8−phenotype, exhibit positivity for the cytotoxic associated antigens TIA-1, granzyme B, and perforin (65, 66). The presence of cytotoxic antigen expression is helpful in the differential diagnosis with HD. In contrast to HD, the cells also are usually positive for leukocyte common antigen. The tumor cells are positive for the interleukin 2 receptor as identified by staining for CD25. Due to loss of many pan T-cell antigens, many cases of ALCL have an apparent “null cell phenotype.” However, no other distinctions can be found in cases with either a T or null cell phenotype. For these reasons, in the Revised European-American Lymphoma and World Health Organization classifications, T/null ALCL is considered a single entity (17, 42).
Early reports of tumors with the features of ALCL found evidence of lysosomal enzyme activity by enzyme histochemistry. The cells were positive for acid phosphatase and nonspecific esterase (67, 68). However, in contrast to the diffuse cytoplasmic reactivity typical of histiocytes, strong perinuclear reactivity was seen (69). This pattern of staining is characteristic of activated lymphoid cells. Granular staining for some antibodies reactive with CD68, such as KP-1, may be seen, but other antibodies, such as PGM1 are negative (70). Antibodies to blood group related antigens (BNH.9) that are negative in HD are generally positive in ALCL (71).
A recent study employing array technology to identify new genes expressed in ALCL identified clusterin as being aberrantly expressed in all cases of systemic ALCL, but not in primary cutaneous ALCL (72). Antibodies to clusterin stained ALCL and follicular dendritic cells, but not other normal T or B lymphocytes. Staining was similarly negative in nearly all non-Hodgkin's lymphomas and HD. One case of T-cell rich large B-cell lymphoma was positive. Clusterin expression might contribute to the propensity of the cells to invade lymphatic sinuses.
The existence of a B-cell form of ALCL has been controversial. Many high grade B-cell lymphomas, especially those associated with EBV, can express CD30 (22, 24, 73). These do not meet strict criteria for a diagnosis of ALCL. There are rare reports of ALCL cases of B-cell origin positive for the t(2;5) by cytogenetics or RT-PCR, or by staining for ALK (44, 74). These have been reported largely from one center, and have not been seen in other series. Further studies may determine whether a B-cell form of ALCL truly exists. Notably, the t(2;5) has been reported by RT-PCR in rare normal peripheral blood specimens. In addition, a rare form of CD30-negative large B-cell lymphoma that is positive for the full length ALK kinase has been described by Delsol et al. (75) This B-cell lymphoma is usually associated with expression of IgA, and is seen in elderly male patients. It has an aggressive clinical course. Because the cells are CD30-negative, confusion with classical ALCL is not a problem. Interestingly, these tumors are usually negative for CD20, the most commonly employed B-cell marker.
GENETIC FEATURES
Immunophenotypic studies may be inconclusive in proving a T-cell origin, but most cases of ALCL show clonal T-cell gene rearrangement at the molecular level (64, 65). Polymerase chain reaction (PCR) techniques for clonal rearrangement of the T-cell receptor gamma and beta chains are usually positive. EBV sequences are absent, although EBV-expressing B-cell lymphomas do express high levels of CD30 and have been misinterpreted as “anaplastic large cell lymphomas” in some older series (76).
The molecular pathogenesis of ALCL has been elucidated in recent years. Since the cloning of the t(2;5)(p23;q32) breakpoint, other variant translocations have been detected (Table 3). They all involve ALK, the ALCL kinase gene on chromosome 2, and other partner genes on chromosomes 1, 2, 3, and 22 (47, 77, 78, 79, 80, 81). Additional partner genes are likely to be identified in the future. All result in upregulation of ALK, but the distribution of the staining varies depending on the translocation. NPM is involved in shuttling of ribosomal proteins into the nucleus (82). The normal function of NPM in the classic t(2;5) leads to positive staining for ALK in both the cytoplasm and nucleus. In the variant translocations, often only cytoplasmic staining will be observed.
Importantly, cases with variant translocations will be negative by RT-PCR, because the RT-PCR technique employs primers that are specific for the ALK and NPM genes (27, 47). However, cases with variant translocations are positive for ALK by immunohistochemistry. Therefore, immunohistochemistry has supplanted molecular tests for the diagnosis of ALCL, based on its sensitivity and specificity. Normal lymphoid cells do not express ALK (5).
Insights into the molecular pathogenesis of ALCL have been important in clarifying the definition of ALCL, and its relationship to other lymphomas including HD. Initially, Stein et al. postulated on the basis of the CD30 expression that ALCL was related to HD (1). In addition, there were morphologic and immunophenotypic overlaps between ALCL and HD that furthered this supposition (41). Although one study found evidence of the NPM/ALK in HD by RT-PCR (18), other reports failed to confirm this observation (19, 20, 21, 83). Similarly, because the morphologic spectrum of ALCL is broad, some studies concluded that the t(2;5) NPM/ALK might not be specific for ALCL (74, 84). Positive results by RT-PCR have been reported in peripheral T-cell lymphomas and “T-immunoblastic lymphomas.” It is likely that these “false-positive” cases were morphologic variants of ALCL, e.g., monomorphic (19), small cell, or lymphohistiocytic variants (33).
CONCLUSION
ALCL has been defined as a distinct clinicopathologic entity based on its histologic, clinical, immunophenotypic, and molecular features. Molecular analysis has been an important tool in clarifying the borderlands of this and other entities, as most diseases have a distinct molecular fingerprint that relates to the underlying pathogenesis. Moreover, in this paradigm molecular studies may be used to refine the ultimate morphologic definition, leading to improved diagnostic criteria. The accurate diagnosis of ALCL has important clinical implications because it is a highly treatable form of lymphoma and has a much better prognosis than other types of T-cell lymphoma. Our current definition of ALCL is both narrower and broader than originally envisioned. A relationship to HD and high grade CD30+ large B-cell lymphomas has been largely disproved. However, ALCL variants such as the small cell variant and the lymphohistiocytic variant have been shown to be part of the same disease entity.
A remaining question for the future is the resolution of the 15 to 20% of cases currently diagnosed as T/null ALCL that are ALK-negative. Based on their clinical and epidemiologic features, it is likely that these ALK-negative cases constitute one or more different entities distinct from ALK-positive ALCL. I propose that such cases would be more appropriately diagnosed as an “anaplastic variant” of peripheral T-cell lymphoma, unspecified. Even if one elects to consider them within the broad definition of ALCL, it is important to designate them as ALK+ or ALK−, because of the significant clinical distinctions. An analogy is the molecular genetic subclassification of acute myeloid leukemia, in which distinctive genetic variants are considered subentities with clinical and prognostic implications (36).
References
Stein H, Mason D, Gerdes J, O'Connor N, Wainscoat J, Pallesen G, et al. The expression of the Hodgkin's disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985; 66: 848–858.
Rimokh R, Magaud JP, Berger F, Samarut J, Coiffier B, Germain D, et al. A translocation involving a specific breakpoint (q35) on chromosome 5 is characteristic of anaplastic large cell lymphoma (“Ki-1 lymphoma”). Br J Haematol 1989; 71: 31–36.
Kaneko Y, Frizzera G, Edamura S, Maseki N, Sakurai M, Komada Y, et al. A novel translocation, t(2;5)(p23;q35), in childhood phagocytic large T-cell lymphoma mimicking malignant histiocytosis. Blood 1989; 73: 806–813.
Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma [published erratum appears in Science 1995 Jan 20;267(5196): 316–7]. Science 1994; 263: 1281–1284.
Pulford K, Lamant L, Morris SW, Butler LH, Wood KM, Stroud D, et al. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood 1997; 89: 1394–1404.
Schwab U, Stein H, Gerdes J, Lemke H, Kirchner H, Schaadt M, et al. Production of a monoclonal antibody specific for Hodgkin and Reed-Sternberg cells of Hodgkin's disease and a subset of normal lymphoid cells. Nature 1982; 299: 65–67.
Stein H, Gerdes J, Schwab U, Lemke H, Mason DY, Ziegler A, et al. Identification of Hodgkin and Sternberg-Reed cells as a unique cell type derived from a newly-detected small-cell population. Int J Cancer 1982; 30: 445–459.
Andreesen R, Osterholz J, Lohr G, Bross K . A Hodgkin cell-specific antigen is expressed on a subset of auto- and alloactivated T (helper) lymphoblasts. Blood 1984; 6: 1299–1302.
Andreesen R, Brugger W, Lohr GW, Bross KJ . Human macrophages can express the Hodgkin's cell-associated antigen Ki-1 (CD30). Am J Pathol 1989; 134: 187–192.
Abbondanzo SL, Sato N, Straus SE, Jaffe ES . Acute infectious mononucleosis. CD30 (Ki-1) antigen expression and histologic correlations [see comments]. Am J Clin Pathol 1990; 93: 698–702.
Durkop H, Latza U, Hummel M, Eitelbach F, Seed B, Stein H . Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin's disease. Cell 1992; 68: 421–427.
Nadali G, Vinante F, Stein H, Todeschini G, Tecchio C, Morosato L, et al. Serum levels of the soluble form of CD30 molecule as a tumor marker in CD30+ anaplastic large-cell lymphoma. J Clin Oncol 1995; 13: 1355–1360.
Nadali G, Tavecchia L, Zanolin E, Bonfante V, Viviani S, Camerini E, et al. Serum level of the soluble form of the CD30 molecule identifies patients with Hodgkin's disease at high risk of unfavorable outcome. Blood 1998; 91: 3011–3016.
Stansfeld A, Diebold J, Kapanci Y, Kelenyi G, Lennert K, Mioduszewska O, et al. Updated Kiel classification for lymphomas. Lancet 1988; i: 292–293.
Lennert K, Feller A . Histopathology of non-Hodgkin's lymphomas. 2nd ed. New York: Springer-Verlag; 1992.
Kadin ME, Sako D, Berliner N, Franklin W, Woda B, Borowitz M, et al. Childhood Ki-1 lymphoma presenting with skin lesions and peripheral lymphadenopathy. Blood 1986; 68: 1042–1049.
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 1994; 84: 1361–1392.
Orscheschek K, Merz H, Hell J, Binder T, Bartels H, Feller AC . Large-cell anaplastic lymphoma-specific translocation (t[2;5] [p23;q35]) in Hodgkin's disease: indication of a common pathogenesis? Lancet 1995; 345 (8942): 87–90.
Wellman A, Otsuki T, Vogelbruch M, Clark HM, Jaffe ES, Raffeld M . Analysis of the t(2;5) (p23;q35) translocation by reverse trasnscription-polymerase chain reaction in CD 30+ anaplastic large-cell lymphomas, in other non-Hodgkin's of T-cell phenotype, and in Hodgkin's disease. Blood 1995; 86: 2321–2328.
Ladanyi M, Cavalchire G, Morris SW, Downing J, Filippa DA . Reverse transcriptase polymerase chain reaction for the Ki-1 anaplastic large cell lymphoma-associated t(2;5) translocation in Hodgkin's disease. Am J Pathol 1994; 145: 1296–1300.
Elmberger PG, Lozano MD, Weisenburger DD, Sanger W, Chan WC . Transcripts of the npm-alk fusion gene in anaplastic large cell lymphoma, Hodgkin's disease, and reactive lymphoid lesions. Blood 1995; 86: 3517–3521.
Falini B, Pileri S, Pizzolo G, Durkop H, Flenghi L, Stirpe F, et al. CD30 (Ki-1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood 1995; 85: 1–14.
Alsabeh R, Medeiros LJ, Glackin C, Weiss LM . Transformation of follicular lymphoma into CD30-large cell lymphoma with anaplastic cytologic features. Am J Surg Pathol 1997; 21: 528–536.
Piris M, Brown D, Gatter K, Mason D . CD30 Expression in non-Hodgkin's lymphoma. Histopathology 1990; 17: 211–218.
Shiota M, Fujimoto J, Takenaga M, Satoh H, Ichinohasama R, Abe M, et al. Diagnosis of t(2;5)(p23;q35)-associated Ki-1 lymphoma with immunohistochemistry. Blood 1994; 84: 3648–3652.
Benharroch D, Meguerian-Bedoyan Z, Lamant L, Amin C, Brugieres L, Terrier-Lacombe MJ, et al. ALK-positive lymphoma: a single disease with a broad spectrum of morphology. Blood 1998; 91: 2076–2084.
Pittaluga S, Wiodarska I, Pulford K, Campo E, Morris SW, Van den Berghe H, et al. The monoclonal antibody ALK1 identifies a distinct morphological subtype of anaplastic large cell lymphoma associated with 2p23/ALK rearrangements. Am J Pathol 1997; 151: 343–351.
Kinney M, Collins R, Greer J, Whitlock J, Sioutos N, Kadin M . A small-cell-predominant variant of primary Ki-1 (CD30)+ T-cell lymphoma. Am J Surg Pathol 1993; 17: 859–868.
Jaffe ES . Post-thymic T-cell lymphomas. In: Jaffe ES, editor. Surgical pathology of the lymph nodes and related organs. 2nd ed. Philadelphia: W.B. Saunders; 1995. p. 344–389.
Bitter MA, Franklin WA, Larson RA, McKeithan TW, Rubin CM, Le Beau MM, et al. Morphology in Ki-1(CD30)-positive non-Hodgkin's lymphoma is correlated with clinical features and the presence of a unique chromosomal abnormality, t(2;5)(p23;q35). Am J Surg Pathol 1990; 14: 305–316.
Nakamura S, Shiota M, Nakagawa A, Yatabe Y, Kojima M, Motoori T, et al. Anaplastic large cell lymphoma: a distinct molecular pathologic entity: a reappraisal with special reference to p80(NPM/ALK) expression. Am J Surg Pathol 1997; 21: 1420–1432.
Pileri S, Falini B, Delsol G, Stein H, Baglioni P, Poggi S, et al. Lymphohistiocytic T-cell lymphoma (anaplastic large cell lymphoma CD30+/Ki1+) with a high content of reactive histiocytes. Histopathol 1990; 16: 383–391.
Pileri SA, Pulford K, Mori S, Mason DY, Sabattini E, Roncador G, et al. Frequent expression of the NPM-ALK chimeric fusion protein in anaplastic large-cell lymphoma, lympho-histiocytic type. Am J Pathol 1997; 150: 1207–1211.
Mann KP, Hall B, Kamino H, Borowitz MJ, Ratech H . Neutrophil-rich, Ki-1-positive anaplastic large-cell malignant lymphoma. Am J Surg Pathol 1995; 19: 407–416.
McCluggage WG, Walsh MY, Bharucha H . Anaplastic large cell malignant lymphoma with extensive eosinophilic or neutrophilic infiltration [see comments]. Histopathology 1998; 32: 110–115.
Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie house, Virginia, November 1997. J Clin Oncol 1999; 17: 3835–3849.
Chan JK, Buchanan R, Fletcher CD . Sarcomatoid variant of anaplastic large-cell Ki-1 lymphoma. Am J Surg Pathol 1990; 14: 983–988.
Rubin BP, Lawrence BD, Perez-Atayde A, Xiao S, Yi ES, Fletcher CDM, et al. TPM-ALK fusion genes and ALK expression in inflammatory myofibroblastic tumor. Mod Pathol 2000; 13: 15A.
Coffin CM, Hussong J, Perkins S, Griffin CA, Perlman EJ . ALK and p80 expression in inflammatory myofibroblastic tumor (IMT). Mod Pathol 2000; 13: 8A.
Pileri S, Bocchia M, Baroni C, Martelli M, Falini B, Sabattini E, et al. Anaplastic large cell lymphoma (CD30+/Ki-1+): results of a prospective clinicopathologic study of 69 cases. Br J Haematol 1994; 86: 513–523.
Leoncini L, Del Vecchio M, Kraft R, Megha T, Barbini P, Cevenini G, et al. Hodgkin's disease and CD30-positive anaplastic large cell lymphomas—a continuous spectrum of malignant disorders. Am J Pathol 1990; 137: 1047–1057.
Jaffe ES, Harris NL, Diebold J, Muller-Hermelink HK . World Health Organization classification of lymphomas: a work in progress. Annu Oncol. 1998; 9 (Suppl. 5): S25–S30.
Falini B, Pileri S, Zinzani PL, Carbone A, Zagonel V, Wolf-Peeters C, et al. ALK+ lymphoma: clinico-pathological findings and outcome. Blood 1999; 93: 2697–2706.
Gascoyne RD, Aoun P, Wu D, Chhanabhai M, Skinnider BF, Greiner TC, et al. Prognostic significance of anaplastic lymphoma kinase (ALK) protein expression in adults with anaplastic large cell lymphoma. Blood 1999; 93: 3913–3921.
Sandlund JT, Pui CH, Roberts WM, Santana VM, Morris SW, Berard CW, et al. Clinicopathologic features and treatment outcome of children with large-cell lymphoma and the t(2;5)(p23;q35). Blood 1994; 84: 2467–2471.
Chan JK, Ng CS, Hui PK, Leung TW, Lo ES, Lau WH, et al. Anaplastic large cell Ki-1 lymphoma. Delineation of two morphological types. Histopathology 1989; 15: 11–34.
Falini B, Pulford K, Pucciarini A, Carbone A, De Wolf-Peeters C, Cordell J, et al. Lymphomas expressing ALK fusion protein(s) other than NPM-ALK. Blood 1999; 94: 3509–3515.
The Non-Hodgkin's Lymphoma Classification Project. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin's lymphoma. Blood 1997; 89: 3909–3918.
Kaudewitz P, Burg G, Stein H . Ki-1 (CD30) positive cutaneous anaplastic large cell lymphomas. Curr Probl Dermatol 1990; 19: 150–156.
de Bruin PC, Beljaards RC, van Heerde P, Van Der Valk P, Noorduyn LA, Van Krieken JH, et al. Differences in clinical behaviour and immunophenotype between primary cutaneous and primary nodal anaplastic large cell lymphoma of T-cell or null cell phenotype. Histopathology 1993; 23: 127–135.
Jaffe ES, Krenacs L, Raffeld M . Classification of T-cell and NK-cell neoplasms based on the REAL classification. Annu Oncol. 1997; 8 (Suppl. 2): S17–S24.
McCarty MJ, Vukelja SJ, Sausville EA, Perry JJ, James WD, Jaffe ES, et al. Lymphomatoid papulosis associated with Ki-1-positive anaplastic large cell lymphoma. A report of two cases and a review of the literature. Cancer 1994; 74: 3051–3058.
Willemze R, Beljaards RC . Spectrum of primary cutaneous CD30 (Ki-1)-positive lymphoproliferative disorders. A proposal for classification and guidelines for management and treatment. J Am Acad Dermatol 1993; 28: 973–980.
DeCoteau JF, Butmarc JR, Kinney MC, Kadin ME . The t(2;5) chromosomal translocation is not a common feature of primary cutaneous CD30+ lymphoproliferative disorders: comparison with anaplastic large-cell lymphoma of nodal origin. Blood 1996; 87: 3437–3441.
Willemze R, Kerl H, Sterry W, Berti E, Cerroni L, Chimenti S, et al. EORTC classification for primary cutaneous lymphomas: a proposal from the Cutaneous Lymphoma Study Group of the European Organization for Research and Treatment of Cancer. Blood 1997; 90: 354–371.
Falini B, Bigerna B, Fizzotti M, Pulford K, Pileri SA, Delsol G, et al. ALK expression defines a distinct group of T/null lymphomas (“ALK lymphomas”) with a wide morphological spectrum. Am J Pathol 1998; 153: 875–886.
Shiota M, Mori S . The clinicopathological features of anaplastic large cell lymphomas expressing p80NPM/ALK. Leuk Lymphoma 1996; 23 (1–2): 25–32.
Fraga M, Brousset P, Schlaifer D, Payen C, Robert A, Rubie H, et al. Bone marrow involvement in anaplastic large cell lymphoma. Immunohistochemical detection of minimal disease and its prognostic significance. Am J Clin Pathol 1995; 103: 82–89.
Pallesen G, Hamilton-Dutoit SJ . Ki-1 (CD30) antigen is regularly expressed by tumor cells of embryonal carcinoma. Am J Pathol 1988; 133: 446–450.
Moller P, Matthaei-Maurer D, Moldenhauer G . CD30(Ki-1) antigen expression in a subset of gastric mucosal plasma cells and in a primary gastric plasmacytoma. Am J Clin Pathol 1989; 91: 18–23.
Penny RJ, Blaustein JC, Longtine JA, Pinkus GS . Ki-1-positive large cell lymphomas, a heterogenous group of neoplasms. Morphologic, immunophenotypic, genotypic, and clinical features of 24 cases. Cancer 1991; 68: 362–373.
Delsol G, Al Saati T, Gatter K, Gerdes J, Schwarting R, Caveriviere P, et al. Coexpression of epithelial membrane antigen (EMA), Ki-1, and interleukin-2 receptor by anaplastic large cell lymphomas: diagnostic value in so-called malignant histiocytosis. Am J Pathol 1988; 130: 59–70.
Herbst H, Tippelmann G, Anagnostopoulos I, Gerdes J, Schwarting R, Boehm T, et al. Immunoglobulin and T. cell receptor gene rearrangements in Hodgkin's disease and Ki-1-positive anaplastic large cell lymphoma: dissociation between phenotype and genotype. Leuk Res 1989; 13: 103–116.
O'Connor N, Stein H, Falini B, Gatter K, Mason D . Genotypic analysis of large cell lymphomas which express the Ki-1 antigen. Histopathology 1987; 11: 733–740.
Foss HD, Anagnostopoulos I, Araujo I, Assaf C, Demel G, Kummer JA, et al. Anaplastic large-cell lymphomas of T-cell and null-cell phenotype express cytotoxic molecules. Blood 1996; 88: 4005–4011.
Krenacs L, Wellmann A, Sorbara L, Himmelmann AW, Bagdi E, Jaffe ES, et al. Cytotoxic cell antigen expression in anaplastic large cell lymphomas of T- and null-cell type and Hodgkin's disease: evidence for distinct cellular origin. Blood 1997; 89: 980–989.
Wilson MS, Weiss LM, Gatter KC, Mason DY, Dorfman RF, Warnke RA . Malignant histiocytosis. A reassessment of cases previously reported in 1975 based on paraffin section immunophenotyping studies. Cancer 1990; 66: 530–536.
Headington J, Roth M, Schnitzer B . Regressing atypical histiocytosis: a review and critical appraisal. Seminars in Diagnostic Pathology 1987; 4: 29–37.
Jaffe ES . Malignant histiocytosis and true histiocytic lymphomas. In: Jaffe ES, editor. Surgical pathology of lymph nodes and related organs. 2nd ed. Philadelphia: W.B. Saunders Co.; 1995. p. 560–593.
Falini B, Flenghi L, Pileri S, Gambacorta M, Bigerna B, Durkop H, et al. PG-M1: a new monoclonal antibody directed against a fixative-resistant epitope on the macrophage-restricted form of the CD68 molecule. Am J Pathol 1993; 142: 1359–1372.
Delsol G, Blancher A, Al Saati T, Ralfkiaer F, Lauritzen A, Bruigeres L, et al. Antibody BNH9 detects red blood cell-related antigens on anaplastic large cell (CD30+) lymphomas. Br J Cancer 1991; 64: 321–326.
Wellmann A, Thieblemont C, Pittaluga S, Sakai A, Jaffe ES, Siebert P, et al. Detection of differentially expressed genes in lymphomas using cDNA arrays: identification of clusterin as a new diagnostic marker for Anaplastic Large Cell Lymphomas (ALCL). Blood 2000; 96: 398–404.
Carbone A, Dolcetti R, Gloghini A, Maestro R, Vaccher E, di Luca D, et al. Immunophenotypic and molecular analyses of acquired immune deficiency syndrome-related and Epstein-Barr virus-associated lymphomas: a comparative study. Hum Pathol 1996; 27: 133–146.
Weisenburger DD, Gordon BG, Vose JM, Bast MA, Chan WC, Greiner TC, et al. Occurrence of the t(2;5)(p23;q35) in non-Hodgkin's lymphoma. Blood 1996; 87: 3860–3868.
Delsol G, Lamant L, Mariame B, Pulford K, Dastugue N, Brousset P, et al. A new subtype of large B-cell lymphoma expressing the ALK kinase and lacking the 2; 5 translocation. Blood 1997; 89: 1483–1490.
Carbone A, Gloghini A, Zanette I, Canal B, Volpe R . Demonstration of Epstein-Barr viral genomes by in situ hybridization in acquired immune deficiency syndrome-related high grade and anaplastic large cell CD30+ lymphomas. Am J Clin Pathol 1993; 99: 289–297.
Wlodarska I, De Wolf-Peeters C, Falini B, Verhoef G, Morris SW, Hagemeijer A, et al. The cryptic inv(2)(p23q35) defines a new molecular genetic subtype of ALK-positive anaplastic large-cell lymphoma. Blood 1998; 92: 2688–2695.
Hernandez L, Pinyol M, Hernandez S, Bea S, Pulford K, Rosenwald A, et al. TRK-Fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally different TFG-ALK translocations. Blood 1999; 94: 3265–3268.
Touriol C, Greenland C, Lamant L, Pulford K, Bernard F, Rousset T, et al. Further demonstration of the diversity of chromosomal changes involving 2p23 in ALK-positive lymphoma: 2 cases expressing ALK kinase fused to CLTCL (clathrin chain polypeptide-like). Blood 2000; 95: 3204–3207.
Ma Z, Cools J, Marynen P, Cui X, Siebert R, Gesk S, et al. Inv(2)(p23q35) in anaplastic large-cell lymphoma induces constitutive anaplastic lymphoma kinase (ALK) tyrosine kinase activation by fusion to ATIC, an enzyme involved in purine nucleotide biosynthesis. Blood 2000; 95: 2144–2149.
Trinei M, Lanfrancone L, Campo E, Pulford K, Mason DY, Pelicci PG, et al. A new variant anaplastic lymphoma kinase (ALK)-fusion protein (ATIC-ALK) in a case of ALK-positive anaplastic large cell lymphoma. Cancer Res 2000; 60: 793–798.
Borer RA, Lehner CF, Eppenberger HM, Nigg EA . Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 1989; 56: 379–390.
Downing JR, Ladanyi M, Raffeld M, Weiss LM, Morris SW . Large-cell anaplastic lymphoma-specific translocation in Hodgkin's disease [letter; comment]. Lancet 1995; 345 (8954): 918.
Downing JR, Shurtleff SA, Zielenska M, Curcio-Brint AM, Behm FG, Head DR, et al. Molecular detection of the (2;5) translocation of non-Hodgkin's lymphoma by reverse transcriptase-polymerase chain reaction. Blood 1995; 85: 3416–3422.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jaffe, E. Anaplastic Large Cell Lymphoma: The Shifting Sands of Diagnostic Hematopathology. Mod Pathol 14, 219–228 (2001). https://doi.org/10.1038/modpathol.3880289
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880289
Keywords
This article is cited by
-
Central nervous system ALK-negative anaplastic large cell lymphoma with IRF4/DUSP22 rearrangement
Brain Tumor Pathology (2022)
-
Hämatologische Neoplasien als Differenzialdiagnosen von Weichgewebstumoren
Der Pathologe (2019)
-
Therapieresistentes Erysipel?
hautnah (2017)
-
Transformation of CLL to ALCL: the role of clonality studies in diagnostic molecular haematopathology
Journal of Hematopathology (2016)
-
Hematopoietic stem cell transplantation in relapsed ALK+ anaplastic large cell lymphoma in children and adolescents: a study on behalf of the SFCE and SFGM-TC
Bone Marrow Transplantation (2015)
