
Abstract
Sinus histocytosis with massive lymphadenopathy, also known as Rosai-Dorfman Disease (RDD), is an idiopathic histiocytic proliferation affecting lymph nodes. Although extranodal involvement has been reported in diverse sites, central nervous system (CNS) manifestation, particularly in the absence of nodal disease is uncommon. We report 11 cases of RDD primary to the CNS without evidence of other sites of involvement. The cases included 7 males and 4 females ranging in age from 22 to 63 years (mean: 41 y). The patients presented with headaches, seizures, numbness, or paraplegia. Eight cases involved the cranial cavity and three cases, the spinal canal. Lesions were most often extra-axial and dura based. Only one presented in the CNS parenchyma. Histologically, the lesions consisted of variable numbers of pale-staining histocytes with emperipolesis often overshadowed by extensive lymphoplasmacytic infiltrates and fibrosis in the background. Special stains for organisms were negative. By immunohistochemical analysis, the characteristic histiocytes were positive for S100 protein and CD68 and negative for CD1a. Treatment consisted of surgical biopsy or excision. Follow-up, available for 10 cases with intervals ranging from 5 days to 42 months (mean: 15 mo), disclosed one patient dying of operative complications 5 days after biopsy and nine patients with no evidence of disease progression RDD should be considered in the differential diagnosis of inflammatory lesions of the CNS. Our study suggests that this entity may have been misdiagnosed in the past as plasma cell granuloma or inflammatory pseudotumor.
Similar content being viewed by others
INTRODUCTION
In 1969, Rosai and Dorfman described a benign histiocytic proliferative disorder that they named sinus histiocytosis with massive lymphadenopathy (SHML, Rosai-Dorfman disease; 1). In its classic form, the disease presents in children and adolescents with massive painless cervical lymphadenopathy, often with associated fever, mild anemia, polyclonal hypergammaglobulinemia, and an elevated sedimentation rate (2). Extranodal Rosai-Dorfman Disease (RDD) has been reported in approximately 43% of cases, either alone or in association with lymphadenopathy. The most common extranodal sites include the skin, orbit, and upper respiratory tract (2); involvement of the central nervous system (CNS), especially in the absence of nodal disease, is rare. The goal of this study was to document the clinicopathologic features of 11 cases of RDD-CNS and to describe criteria for distinguishing this disorder from similar-appearing infectious and from neoplastic and nonspecific reactive conditions.
MATERIALS AND METHODS
A search of the files of the Neuropathology Registry at the Armed Forces Institute of Pathology (Washington, DC) from 1977 to the present disclosed 11 cases of RDD-CNS, one of which has previously been reported by the original contributors (3, 4). Hematoxylin and eosin–stained slides and a clinical history were available for each case; radiographic studies were reviewed in one case. Clinical follow-up was obtainable for 10 of 11 cases. Special stains for organisms including Grocott methenamine silver, Ziehl-Neelsen, Brown-Hopps Gram stain, periodic acid-Schiff, and Warthin Starry were performed in eight cases. Immunoperoxidase evaluation using the avidin-biotin-complex method of Hsu (5) was performed using a panel of antibodies (Table 1). Enzyme enhancement involved 0.04% pepsin at pH 2.0 for 20 minutes at 40°C. Microwave antigen retrieval involved heating sections in a microwave oven in 1 mmol/L EDTA buffer solution at pH 8.0 for 10 minutes.
RESULTS
Clinical Findings
The clinical data is summarized in Table 2. There were seven males and four females, ranging in age from 22 to 63 years (mean age: 42 y). The eight patients with intracranial lesions came to medical attention because of headaches, seizures, or visual loss. The three patients with spinal disease presented with spastic paraparesis or paraplegia. All except one case manifested extraparenchymal disease associated with the dura. Case 8, which was previously reported (3, 4), presented with an intramedullary spinal cord lesion unassociated with any extramedullary component. None of the patients was febrile or demonstrated lymph node involvement either at presentation or subsequently. One patient disclosed mild normochromic, normocytic anemic and polyclonal hypergammaglobulinemia on preoperative evaluation. An elevated erythrocyte sedimentation rate was detected in two of the three cases in which this test was performed. The most common clinical diagnosis was meningioma, which was the working preoperative diagnosis in seven of eight patients with intracranial disease.
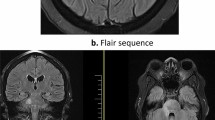
Magnetic resonance imaging studies from Case 9 disclosed a lesion involving the dura at the right orbital apex and cavernous sinus region (Fig. 1). The lesion displayed slightly hyperintense signal in T1W images, enhanced markedly after gadolinium administration, and showed hypointensity on T2W studies. Magnetic resonance image studies from Case 11 showed a 5 × 1-cm extra-axial mass along the occipital bone that enhanced uniformly after gadolinium administration.

Magnetic resonance image of Case 9 showing enhancing extra-axial mass in the right parasellar region.
Pathologic Findings
Histologically, all 11 cases showed nodular cellular infiltrates composed of histiocytes mixed with plasma cells and lymphocytes in a background of increased collagen and/or reticulin fibers (Fig. 2). The typical histiocyte showed indistinct cellular margins and contained abundant cytoplasm, which was clear to eosinophilic and finely or coarsely vacuolated. The nuclei were round or oval and demonstrated a vesicular chromatin pattern and a single small nucleolus. Grooved or lobulated nuclei were not prominent. Occasional histiocytes were multinucleated. Unequivocal emperipolesis could be identified in hematoxylin and eosin–stained sections in seven cases (Fig. 3). Examination of immunostained sections facilitated the recognition of emperipolesis in the remaining four cases. The phagocytized cells were most often lymphocytes, but plasma cell and erythrocytes could also be occasionally seen engulfed within the histiocyte cell cytoplasm.

Nodular infiltrates of pale-staining histiocytes and chronic inflammatory cells in a fibrotic background.

A characteristic histiocyte showing emperipolesis of small lymphocytes.
The lymphocytic and plasma cell infiltrates were composed of mature cells (Fig. 4). In several cases, the nonhistocytic infiltrates obscured the histiocytic population. Binucleate plasma cells and intracytoplasmic eosinophilic inclusions (Russell bodies) were commonly observed. Blood vessels were frequently outlined by cuffs of plasma cells and/or lymphocytes. Necrosis, well-formed granulomas, and eosinophilia were consistently absent. Microorganisms were not detected in any of the cases examined.

Scattered large histiocytes intermixed with numerous small lymphocytes and plasma cells.
The characteristic histiocytes were strongly positive for S-100 protein and CD68 (Fig. 5). Seven cases were negative when examined for CD1a. Staining for CD20 and CD3 showed a mixed population of B and T lymphocytes in the background. Kappa and lambda light immunohistochemistry showed polytypic staining in the plasma cell infiltrates. The stain for EMA decorated plasma cells but no other cellular constituents in the three cases tested.

Numerous histiocytes showing S100 immunoreactivity.
Treatment and Follow-Up
All patients underwent surgery with biopsy, subtotal excision, or resection of the symptom producing mass. None of the patients were treated with radiation or chemotherapy. One patient died from surgical complications 5 days after craniotomy. Follow-up intervals in 9 of the 10 remaining patients ranged from 2 to 42 months, with a mean of 15 months. In cases with complete local excision, there has been no evidence of recurrence, and in cases with subtotal resection, clinical or radiographic progression of disease has not been observed.
DISCUSSION
With an estimated incidence of approximately 100 cases per year in the United States, RDD is a rare condition, even in its classic nodal presentation. Although extranodal disease is seen in up to 43% of cases, CNS involvement characterizes less than 5% of the patient total (2, 6). Of the 37 previous cases of RDD-CNS, 26 were intracranial (2, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22), eight involved the spine (2, 3, 4, 23, 24, 25, 26), and three had both spinal and intracranial disease (12, 18). Most intracranial tumors were associated with the dura, thereby mimicking meningioma both clinically and radiographically. Aside from Case 8, which had an unusual intramedullary location, previous cases of spinal RDD were likewise dura-based masses. RDD-CNS shows a predilection for males and typically presents during the fourth to fifth decade, in contrast to nodal-based RDD, with a mean age of presentation of 20.6 years (2, 9). The symptoms at presentation reflect the site of disease and include seizures, headaches, cranial nerve deficits, and paraparesis. Rare cases with sella involvement have presented with symptoms related to pituitary dysfunction (12, 15). Although none of the cases in this series had lymph node involvement or other sites of involvement, approximately half of the previously reported cases had disease outside of the CNS, most commonly cervical lymph nodes, skeletal system, or salivary gland (2, 11).
The histologic features of RDD-CNS are similar to those of lymph nodes. Cytologically, the infiltrates are composed of variable numbers of histiocytes intermixed with plasma cells and lymphocytes. As illustrated by the contributor diagnoses in our cases, the diagnosis of RDD-CNS often presents difficulties. Emperipolesis, a consistent finding in nodal disease, is often less apparent in extranodal sites (2) and could be confidently identified in only 7 of 11 cases by hematoxylin and eosin staining morphology. Fibrosis, another feature commonly seen in extranodal sites, was prominent in all cases and often obscured the histiocytic proliferation. S100 protein immunostaining dramatically increased the visibility of the histiocytes, making their true proportion in the cellular infiltrate readily apparent.
Histologic differential diagnostic considerations include several hematopoietic and primary CNS lesions. Lymphoplasmacytic meningioma elicits a pronounced chronic inflammatory reaction, which may be confused with the infiltrates of RDD-CNS (27). Recognition of typical meningioma histology coupled with confirmatory immunostaining for epithelial membrane antigen readily differentiates these two processes. Langerhans histiocytosis often presents as discrete nodules attached to the dura and may exhibit a xanthomatous appearance microscopically. Cytologically, the nuclei of Rosai-Dorfman histiocytes lack the lobation and longitudinal nuclear grooves characteristic of Langerhans cells. Eosinophils, often prominent in Langerhans histiocytoses, are not conspicuous in the cellular infiltrates of RDD. Although RDD and Langerhans' cell histiocytosis are both immunoreactive with S100, CD1a is usually negative in SHML (28, 29). Because of the presence of fibrosis, RDD-CNS may have a distinctly nodular appearance suggestive of the nodular sclerosing variant of Hodgkin's disease. Hodgkin's disease occurring in the CNS is extremely rare and is typically associated with relapse (30). Although classic Reed Sternberg cells are readily distinguished from RDD histiocytes, mononuclear variants, particularly lacunar cells may mimic SHML histiocytes. Classic Reed Sternberg cells and variants, however, lack emperipolesis and S100 immunoreactivity and are typically positive for CD15 and CD30. Intracranial involvement by plasmacytoma is also rare but well documented (31, 32). Similar to the case of RDD-CNS, the lesions are attached to the dura and radiographically resemble meningioma. This diagnosis is easily excluded by demonstrating that the plasma cell infiltrates of RDD-CNS are polyclonal.
As described in the literature, plasma cell granuloma is a discrete, dura-based inflammatory mass with associated fibrosis (33, 34, 35, 36). We believe that many lesions previously reported as CNS plasma cell granulomas or inflammatory pseudotumors are, in fact, RDD-CNS. Mirra et al. raised this possibility as early as 1983 (37), and the premise was echoed by Song (21) and by Burger and Scheithauer (38). Several historic and current case reports of CNS plasma cell granuloma make no mention of the histiocytic contribution, if any, to the lesion's cellular infiltrate, and they omit immunostaining for S-100 and histiocytic markers from the pathologic investigation. Of note, the accompanying pathologic illustrations show characteristic architectural and cytologic features of RDD, including, in some instances, unrecognized emperipolesis. We recommend that before rendering a diagnosis of plasma cell granuloma, RDD-CNS should be excluded through careful evaluation for emperipolesis and S100 immunostaining.
Follow-up data was available for 43 patients, including 10 of 11 patients in this series. Interestingly, this site of involvement did not adversely affect prognosis. The majority of patients (58%) were alive with persistent disease, whereas only two patients (4.7%) had died of RDD-CNS or related surgical complications. Although a variety of treatment modalities have been used including steroid therapy and radiation, surgical resection appears to be the most efficacious approach (8, 10).
The etiology of SHML/RDD is not well understood. The immunophenotypic profile and studies of monokine expression suggest derivation from activated macrophages, which have been shown to produce interleukin-Iβ and tumor necrosis factor-α (39). Although an infection has been suggested as an underlying cause, a definitive agent has never been isolated (2). Molecular studies using polymorphic regions of the human androgen receptor locus have demonstrated that RDD is a polyclonal disorder (40), in contrast to the Langerhans cell histiocytosis, which is monoclonal (41).
In conclusion, we summarize our experience derived from 11 cases of RDD-CNS, 10 of which have not been previously reported. Although rare in the CNS, RDD typically presents as a dura-based mass in the cranial cavity or spinal cord that may or may not be associated with nodal disease. This disorder has probably been underdiagnosed in the past and should be included in the differential diagnosis of fibrotic chronic inflammatory lesions of the CNS.
References
Rosai J, Dorfman RF . Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol 1969; 87: 63–70.
Foucar E, Rosai J, Dorfman R . Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol 1990; 7: 19–73.
Jones MP, Rueda-Pedraza ME . Extranodal sinus histiocytosis with massive lymphadenopathy presenting as an intramedullary spinal cord tumor: a case report. Am J Hematol 1997; 54: 253–257.
Osenbach RK . Isolated extranodal sinus histiocytosis presenting as an intramedullary spinal cord tumor with paraplegia. Case report. J Neurosurg 1996; 85: 692–696.
Hsu S-M, Raine L, Fanger G . Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem 1981; 29: 577–580.
Foucar E, Rosai J, Dorfman RF, Brynes RK . The neurologic manifestations of sinus histiocytosis with massive lymphadenopathy. Neurology 1982; 32: 365–372.
Huang HY, Huang CC, Lui CC, Chen HJ, Chen WJ . Isolated intracranial Rosai-Dorfman disease: case report and literature review. Pathol Int 1998; 48: 396–402.
Kim M, Provias J, Bernstein M . Rosai-Dorfman disease mimicking multiple meningioma: case report. Neurosurgery 1995; 36: 1185–1187.
Deodhare SS, Ang LC, Bilbao JM . Isolated intracranial involvement in Rosai-Dorfman disease: a report of two cases and review of the literature. Arch Pathol Lab Med 1998; 122: 161–165.
Udono H, Fukuyama K, Okamoto H, Tabuchi K . Rosai-Dorfman disease presenting multiple intracranial lesions with unique findings on magnetic resonance imaging. Case report. J Neurosurg 1999; 91: 335–339.
Sakai K, Koike G, Seguchi K, Nakazato Y . Sinus histiocytosis with massive lymphadenopathy: a case of multiple dural involvement. Brain Tumor Pathol 1998; 15: 63–69.
Kelly WF, Bradey N, Scoones D . Rosai-Dorfman disease presenting as a pituitary tumour. Clin Endocrinol (Oxf) 1999; 50: 133–137.
Resnick DK, Johnson BL, Lovely TJ . Rosai-Dorfman disease presenting with multiple orbital and intracranial masses. Acta Neuropathol 1996; 91: 554–557.
Panicker NK, Sabhikhi AK, Rai R . Rosai-Dorfman disease presenting as a meningioma. Indian J Cancer 1996; 33: 192–194.
Ng HK, Poon WS . Sinus histiocytosis with massive lymphadenopathy localized to the sella. Br J Neurosurg 1995; 9: 551–555.
Katz DS, Poe LB, Corona RJ Jr . Sinus histiocytosis with massive lymphadenopathy: a case of simultaneous upper respiratory tract and CNS disease without lymphadenopathy. AJNR Am J Neuroradiol 1993; 14: 219–222.
Shaver EG, Rebsamen SL, Yachnis AT, Sutton LN . Isolated extranodal intracranial sinus histiocytosis in a 5-year-old boy. Case report. J Neurosurg 1993; 79: 769–773.
Foucar E, Rosai J, Dorfman RF, Brynes RK . The neurologic manifestations of sinus histiocytosis with massive lymphadenopathy. Neurology 1982; 32: 365–372.
Haas RJ, Helmig MS, Meister P . Sinus histiocytosis with massive lymphadenopathy and epidural involvement. Hamatol Bluttransfus 1981; 27: 239–243.
Bhattacharjee MB, Wroe SJ, Harding BN, Powell M . Sinus histiocytosis with massive lymphadenopathy–isolated suprasellar involvement. J Neurol Neurosurg Psychiatry 1992; 55: 156–158.
Song SK, Schwartz IS, Strauchen JA, et al. Meningeal nodules with features of extranodal sinus histiocytosis with massive lymphadenopathy. Am J Surg Pathol 1989; 13: 406–412.
Morandi X, Godey B, Riffaud L, Heresbach N, Brassier G . Isolated Rosai-Dorfman disease of the fourth ventricle. J Neurosurg 2000; 92: 890–.
Kessler E, Srulijes C, Toledo E, Shalit M . Sinus histiocytosis with massive lymphadenopathy and spinal epidural involvement: a case report and review of the literature. Cancer 1976; 38: 1614–1618.
Rosai J, Dorfman RF . Sinus histiocytosis with massive lymphadenopathy and spinal epidural involvement: a case report and review of the literature. Cancer 1976; 38: 1614–1618.
Haas RJ, Helmig MS, Prechtel K . Sinus histiocytosis with massive lymphadenopathy and paraparesis: remission with chemotherapy. A case report. Cancer 1978; 42: 77–80.
Chan KW, Chow YY, Ghadially FN, Stansfeld AG, Woo CH . Rosai-Dorfman disease presenting as spinal tumor. A case report with ultrastructural and immunohistochemical studies. J Bone Joint Surg 1985; 67: 1427–1431.
Horten BC, Urich H, Stefoski D . Meningiomas with conspicuous plasma cell-lymphocytic components: a report of five cases. Cancer 1979; 43: 258–264.
Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol 1997; 29: 157–166.
Lopez P, Estes ML . Immunohistochemical characterization of the histiocytes in sinus histiocytosis with massive lymphadenopathy: analysis of an extranodal case. Hum Pathol 1989; 20: 711–715.
Sapozink MD, Kaplan HS . Intracranial Hodgkin's disease. A report of 12 cases and review of the literature. Cancer 1983; 52: 1301–1307.
Krumholz A, Weiss HD, Jiji VH, Bakal D, Kirsh MB . Solitary intracranial plasmacytoma: two patients with extended follow-up. Ann Neurol 1982; 11: 529–532.
Mancardi GL, Mandybur TI . Solitary intracranial plasmacytoma. Cancer 1983; 51: 2226–2233.
West SG, Pittman DL, Coggin JT . Intracranial plasma cell granuloma. Cancer 1980; 46: 330–335.
Le Marc'hadour F, Fransen P, Labat-Moleur F, Passagia JG, Pasquier B . Intracranial plasma cell granuloma: a report of four cases. Surg Neurol 1994; 42: 481–488.
Eimoto T, Yanaka M, Kurosawa M, Ikeya F . Plasma cell granuloma (inflammatory pseudotumor) of the spinal cord meninges: report of a case. Cancer 1978; 41: 1929–1936.
Makino K, Murakami M, Kitano I, Ushio Y . Primary intracranial plasma-cell granuloma: a case report and review of the literature. Surg Neurol 1995; 43: 374–378.
Mirra SS, Tindall SC, Check IJ, Brynes RK, Moore WW . Inflammatory meningeal masses of unexplained origin. An ultrastructural and immunological study. J Neuropathol Exp Neurol 1983; 42: 453–468.
Burger PC, Scheithauer BW . Tumors of the Central Nervous System. Washington, D.C.: Armed Forces Institute of Pathology, 1993;
Foss HD, Herbst H, Araujo I, et al. Monokine expression in Langerhans' cell histiocytosis and sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease) [see comments]. J Pathol 1996; 179: 60–65.
Paulli M, Bergamaschi G, Tonon L, et al. Evidence for a polyclonal nature of the cell infiltrate in sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease). Br J Haematol 1995; 91: 415–418.
Willman CL, Busque L, Griffith BB, et al. Langerhans'-cell histiocytosis (histiocytosis X)–a clonal proliferative disease [see comments]. N Engl J Med 1994; 331: 154–160.
Author information
Authors and Affiliations
Additional information
The opinions and assertions contained herein are the private views of the authors and should not be construed as official or as reflecting the views of the Departments of Army, Navy, or Defense.
Rights and permissions
About this article
Cite this article
Andriko, JA., Morrison, A., Colegial, C. et al. Rosai-Dorfman Disease Isolated to the Central Nervous System: A Report of 11 Cases. Mod Pathol 14, 172–178 (2001). https://doi.org/10.1038/modpathol.3880278
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880278
Keywords
This article is cited by
-
Neurological Manifestations of Histiocytic Disorders
Current Neurology and Neuroscience Reports (2023)
-
Surgical management and outcome of primary intracranial Rosai-Dorfman disease: a single-institute experience and pooled analysis of individual patient data
Neurosurgical Review (2023)
-
Ophthalmic Rosai–Dorfman disease: a multi-centre comprehensive study
BMC Ophthalmology (2021)
-
Diagnosis and treatment of Rosai-Dorfman disease of the spine: a systematic literature review
Systematic Reviews (2021)
-
Rosai–Dorfman Disease: A Rare Clinicopathological Presentation and Review of the Literature
Head and Neck Pathology (2021)
