
Abstract
Six patients had blood and bone marrow manifestations characterized by the presence of morphologically immature or blastic B-lineage lymphoid cells expressing CD5 antigen. The median patient age was 70 years, and the male-to-female ratio was 5:1. The presence or degree of lymphadenopathy and splenomegaly was variable among this group at staging evaluation, although two patients did not have these features. One patient had an antecedent diagnosis of classical nodal mantle cell lymphoma, without prior morphologic blood or bone marrow involvement. Other patients lacked a history of underlying lymphoproliferative disorders. The median white blood cell count was 120 × 109/L. Most patients had thrombocytopenia, whereas only one patient had neutropenia at presentation. Leukemic peripheral blood cells in these six cases were small to medium in size with fine or granular nuclear chromatin and small or inconspicuous nucleoli. The pattern of marrow involvement was interstitial or diffuse, with cells showing immature nuclear features resembling acute leukemia or blastic lymphoma. All tumors demonstrated a consistent immunophenotype of B-cell lineage, surface immunoglobulin positivity, and CD5 antigen expression. The progenitor cell-associated markers CD34 and TdT were not expressed, and CD23 antigen was either negative (three of four cases) or only weakly present (one of four cases). The presence of a karyotypic t(11;14)(q13;q32) was documented in one tumor, whereas two other cases had BCL-1 gene rearrangements by either polymerase chain reaction or Southern blot analysis. Cyclin D1 mRNA overexpression was noted in three of four cases tested. This patient group was characterized by very poor overall survival (median, 3 months; range, 0.5 to 6 months). The aggregate clinical, pathologic, and genetic data in these unusual cases are consistent with de novo or predominant leukemic presentations of blastic mantle cell lymphoma. Accurate diagnosis in such cases is greatly facilitated by cytogenetic studies or the demonstration of BCL-1/cyclin D1 abnormalities.
Similar content being viewed by others

INTRODUCTION
The expression of surface immunoglobulin (SIg) generally signifies a mature B-cell phenotype and is usually accompanied by nuclear changes of chromatin condensation. However, exceptional lymphoid neoplasms characterized by blastic cytology and mature phenotype have been described. One example is a previous report of nine cases of “mature” B-cell acute lymphoblastic leukemia (ALL), characterized by non-L3 leukemic blast morphology and the presence of SIg positivity (1). These cases were phenotypically somewhat heterogeneous, although TdT was consistently negative in each case and CD10 expression was observed in all of five tumors tested for this antigen. Among these unusual blastic, SIg-positive leukemic cases, karyotypic analysis also revealed heterogeneity. Two tumors exhibited 8(q24) region translocations, implying C-MYC gene deregulation. Of interest, one of these lesions had a t(14;18) anomaly, in addition to 8(q24) rearrangement, suggesting blastic transformation of an underlying low grade lymphoproliferative disorder (e.g., follicular lymphoma). It is interesting that two of five tumors in this series were noted to have CD5 coexpression.
In the present study, six cases of B-cell leukemia with a “mature” immunophenotype characterized by SIg positivity, CD5 expression, and lack of CD34 and Tdt are described. The initial blood or bone marrow (BM) morphologic presentation in each case suggested the diagnosis of ALL. The typical cytologic features of chronic lymphocytic leukemia (CLL), prolymphocytic leukemia (PLL), or classical peripheralizing mantle cell lymphoma (MCL) were not evident. Notable is that further evaluation revealed variable extramedullary dissemination in some of these patients. The primary or predominant involvement of peripheral blood (PB) and BM, along with immunophenotypic and genetic findings, is consistent with florid leukemic disease presentations of blastic MCL.
MATERIALS AND METHODS
Case Selection and Clinical Data
Cases of blastic CD5-positive B-cell leukemias characterized by mature immunophenotype (i.e., SIg positivity) were identified from the consultation files of the hematopathology services of the University of New Mexico Hospital and British Columbia Cancer Agency. Wright or May-Grunwald-Giemsa stains of PB and BM samples were reviewed. BM core biopsies were fixed in B5 and decalcified, and 3- to 5-μm sections were stained with hematoxylin and eosin. Pretreatment clinical and staging information, therapeutic details, and outcome data were obtained.
Flow Cytometry and Immunoperoxidase
Standard two- or three-color flow cytometric immunophenotyping was performed on Ficoll-Hypaque (Sigma, St. Louis, MO) processed or red cell lysed cell suspensions of fresh or viably cryopreserved blood and BM specimens. Antibodies used in this study included those against B-cell–related antigens (CD19, CD20, CD22, CD23, κ, and λ) T-cell antigens (CD2, CD3, CD4, CD5, CD7, and CD8), myeloid antigens (CD13, and CD33), and others (CD10, CD34, FMC-7, HLA-DR, and TdT). All antibodies were direct conjugates and were obtained from Becton-Dickinson (San Jose, CA) or Immunotech (Westbrook, ME). Samples were run on FACSCAN (Becton-Dickinson) or Elite XL (Coulter Corp., Miami, FL) cytometers, using appropriate gating techniques. Analysis was carried out using LYSIS II, Paint-a-Gate (Becton-Dickinson), or Winlist v4.0 (Verity Software House, Topsham, ME) software. In the single case in which flow cytometry was not performed in-house (Patient PN4), the phenotyping data were obtained and reviewed.
Immunoperoxidase markers for CD34 and TdT were performed on a Ventana automated immunostainer (Ventana Medical Systems, Tucson, AZ) following standard citrate buffer microwave antigen retrieval, using the appropriate proprietary secondary antibodies and 3,3′-diaminobenzidine as chromogen. All cases were concurrently run with positive and negative controls.
Cytogenetics
Twenty-four- or 48-h unstimulated blood samples were routinely processed for cytogenetics using standard G-banding technique with trypsin-Giemsa preparation. Interpretation was carried out according to the International System for Human Cytogenetic Nomenclature, 1995.
Molecular Analyses
When fresh or frozen material was available, high-molecular-weight DNA was obtained using the Puregene kit (Gentra, Minneapolis, MN), according to the manufacturer’s directions. Total RNA was isolated in two cases using the RNAzol B method (Teltest Inc., Friendswood, TX). For immunoglobulin rearrangements, DNA polymerase chain reaction (PCR) was carried out using a consensus VH FRIII-JH primer strategy. BCL-1 locus rearrangements were assessed with a consensus BCL-1 major translocation cluster (MTC) region primer and the same JH primer, as previously described (2, 3). DNA integrity was verified by amplification of a segment of the β-globin gene. Primer sequences are as follows (all 5′-3′): VH-TGTCGACACGGC(CT)(GC)TGTATTACTG; JH-ACCTGAGGAGACGGTGACC; BCL-1 MTC-GATGGGCTTCTCTCACCTACTA; β-globin I-GGTTGGCCAATCTACTCCCAGG; β-globin II-GCTCACTCAGTGTGGCAAAG. PCR was performed from 1 ug DNA under the following conditions: 95° C × 3 min, 2 cycles of 95° C × 30 seconds, 61° C × 15 seconds, 72° C × 30 seconds, 30 cycles of 95° C × 15 seconds, 58° C × 30 seconds, 72° C × 30 seconds for VH/JH PCR; and 95° C × 3 min, 30 cycles of 95° C × 15 seconds, 57° C × 15 seconds, 72° C × 30 seconds for BCL-1/JH PCR. PCR was followed by a 5-min extension at 72° C in either case. Standard PCR buffer containing 10 mm Tris (pH 8.4), 50 mm KCl, 1.5 mm MgCl2, and 0.2 mm dNTP was used, with 15 to 20 pmol of each primer and 0.5u Taq polymerase (Perkin-Elmer Roche, Branchburg, NJ). PCR products were analyzed on a 10% polyacrylamide gel (ReadyGel and Mini-Protean II system; BioRad, Hercules, CA) for VH/JH or a 1.5% agarose gel (FMC Bioproducts, Seakem, ME) for BCL-1/JH, stained with ethidium bromide and illuminated under ultraviolet light. BCL-1/JH PCR products were subsequently transferred to a nylon membrane (Pall-Biodyne, East Hills, NY) and probed with a biotinylated MTC oligonucleotide sequence (5′-3′: TCAGGCCTTGATAGCTCG) (2, 3) using the Amersham ECL chemiluminescence kit (Amersham Life Sciences, Arlington Heights, IL).
Southern blot (SB) hybridization analyses for JH and BCL-1 gene rearrangements were performed using standard procedures. For the JH locus, DNA was digested with EcoRI and BamHI (Promega, Madison, WI), and for BCL-1, these two enzymes plus SacI or HindIII were used. Probes were labeled with P32 (New England Nuclear, Boston, MA) to high specific activity and included a 2.2 kb JH cDNA (Oncogene Sciences, Uniondale, NY) and a 2.1 kb SacI BCL-1 MTC genomic fragment (courtesy of Dr. Y. Tsujimoto). Blots were exposed on Kodak XAR film (Eastman-Kodak, Rochester, NY) for 2 to 14 days at −70° C.
Cyclin D1 mRNA expression was determined by a qualitative reverse transcriptase (RT)-PCR assay. Amplification of a segment of β2-microglobulin was used as an RNA integrity control. One ug of RNA was reverse transcribed to cDNA using Maloney murine leukemia virus–reverse transcriptase-RT (Gibco-BRL, Grand Island, NY) in a buffer containing 10 mm Tris (pH 8.4), 50 mm KCl, 5.0 mm MgCl2, 5 mm DTT, 1 unit RNAsin (Promega) and 50 pmol random hexamers. PCR was then carried out as follows: 95° C × 5 min, 30 cycles of 95° C × 45 seconds, 58° C × 30 sec, 72° C × 60 seconds and final extension of 72° C × 5 min. Primer sequences are as follows (5′-3′): Cyclin D1 sense-GAGGCGGAGGAGAACAAACA; Cyclin D1 antisense-GAAGCGTGTGAGGCGGTAG; β2 I-GAAAAAGATGAGTATGCCTG; β2 II-ATCTTCAAACCTCCATGATG (β2 microglobulin control). PCR products were analyzed in a 1.5% agarose gel as described above. A case of MCL with a known BCL-1 MTC rearrangement was used as positive control. A prominent PCR product band relative to the positive control was considered positive for cyclin D1 mRNA overexpression. The sequence of cyclin D1 cDNA was obtained from GenBank (Account no. X59798, X59485).
RESULTS
Clinical Features
The clinical, hematologic, morphologic, treatment, and outcome features of these cases are presented in Table 1. Patients were elderly, with a median age of 70 years (range, 55 to 94). The male-to-female ratio was 5:1. One patient (PN2) had an antecedent history of mediastinal and axillary nodal MCL (treated with CHOP chemotherapy) established nearly 3 years before developing blastic leukemic disease. Of note, this patient had a normal staging marrow at initial diagnosis and a negative marrow examination only 1 year before leukemic presentation, despite persistent nodal MCL of classical (nonblastic) histologic type. Patients PN1 and PN5 had normal complete blood cell count and differential studies noted 4 and 6 months before their respective disease occurrences. Clinical evaluation of patients at diagnosis generally revealed absence of significant peripheral adenopathy, with one patient (PN6) demonstrating small bilateral cervical lymph nodes. Patients PN1 and PN3 had additional radiographic studies that did not reveal sites of significant deep-seated adenopathy; however, Patient PN5 was found to have evidence of intra-abdominal and mediastinal adenopathy by CT scan. Splenomegaly was present in Patients PN1 and PN5. Patient PN2 had at diagnosis a paraspinal mass, which was not biopsied. The median white blood cell count at presentation was 120 × 109/L (range, 40 to 199 × 109/L). Anemia was variably present; however, five of six patients had mild to moderate thrombocytopenia. Absolute neutropenia was observed in only one of six cases. Patients received individualized therapy, including palliative care, standard adult ALL-type induction or adriamycin-based, curative intent chemotherapy for intermediate grade lymphoma. Disease-free survival and overall survival were poor (median overall survival, 3 months; range, 0.5 to 6 months).
Morphologic Findings
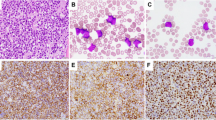
The light microscopic characteristics of these cases are summarized in Table 2. Paired PB and BM specimens were assessed in four of six cases; two cases (PN1, PN2) had only PB available as BM specimens were not obtained. Peripheral smear examination in all six cases revealed circulating immature cells and prompted an initial consideration of acute leukemia (Fig. 1A–C). These cells were characterized by small to medium size, high nuclear:cytoplasmic ratio, dispersed fine or granular chromatin, and relatively small or inconspicuous nucleoli. Extensive BM infiltrates were interstitial or diffuse in the four cases with specimens obtained, showing similar blastic cells with variable nuclear irregularity (Fig. 1D, E). Notable is that in none of these cases did the blastic cells resemble ALL-L3 or Burkitt’s-type cells. Cytoplasmic granularity and Auer rods were not observed, and special stains, including Sudan black B and nonspecific esterase, were negative in two cases examined. Finally, none of these lymphoid tumors cytologically resembled classical peripheralizing MCL, CLL, atypical CLL, or PLL in the blood.

Representative peripheral blood smears of PN1 (A), PN2 (B), and PN5 (C) showing blastic circulating cells with high nuclear:cytoplasmic ratio resembling acute lymphoblastic leukemia. Cells from PN2 show fine chromatin structure, whereas cells from PN1 and PN5 have more granular but dispersed chromatin. D, E, bone marrow biopsy of PN5 showing diffuse replacement of marrow and infiltration by blastic, irregular cells.
Immunophenotypic Analysis
All cases in this series demonstrated consistent immunophenotypic features (Table 3), including pan-B cell Ag expression (CD19, CD20), moderately bright monotypic SIg light chain, HLA-DR, and coexpression of CD5. Lambda light chain–restricted cases were twice as commonly encountered. CD23 expression was absent in three of four cases and was only weakly positive in the remaining leukemia. Markers of immaturity were not present by either flow cytometry or tissue immunoperoxidase; all six cases were CD34 negative, and two of two cases also lacked TdT.
Cytogenetics and Molecular Analyses
Results of cytogenetic and molecular investigations are summarized in Table 3. In one patient (PN1), a der t(11;14)(q13;q32) was observed. Other abnormalities in this case included del (6)(q13), commonly encountered in aggressive lymphoid neoplasms, and a der t(9;14) involving 9(p11) and the p arm of the rearranged chromosome 14. Patient PN5 had a normal male karyotype, although only 13 metaphases were analyzed.
DNA PCR analysis demonstrated clonal IgH rearrangements in all five cases with available material, and this was confirmed by SB analysis of the JH locus in two cases (data not shown). DNA PCR analysis for MTC-associated BCL-1/IgH fusions was positive in one of five cases tested (PN5, Fig. 2), with verification of amplifiable control DNA in the appropriate size range in all samples examined (data not shown). One of three cases (PN3) analyzed for BCL-1 MTC locus rearrangements by SB technique was positive using the MTC region probe (data not shown). However, because fewer than half of SB- or PCR-detectable cases of MCL involve the MTC locus (4, 5, 6), we cannot exclude the possibility of non-MTC–related BCL-1 breakpoint events in the remaining cases analyzed by these molecular methods.

BCL-1/JH PCR analysis. Chemiluminescent hybridization blot of BCL-1/JH polymerase chain reaction (PCR) product from Patient PN5 tumor sample demonstrating presence of a BCL-1 major translocation cluster gene rearrangement. The PCR product was hybridized with an internal BCL-1 sequence oligoprobe. Other lanes: +C, positive control (lymph node involved by classical mantle cell lymphoma); −, negative control (no DNA). A BCL-1 gene rearrangement was also demonstrated in the tumor from patient PN3 using Southern blot technique (not shown). Similarly analyzed samples (by PCR or Southern blot) from Patients PN1, PN2, and PN6 did not reveal BCL-1 major translocation cluster locus rearrangements (see Table 3).
Four patients had sufficient material for cyclin D1 RT-PCR analysis. Three of these four leukemias (PN1, PN5, PN6) demonstrated clear cyclin D1 overexpression in the RT-PCR assay (Fig. 3). Unexpectedly, the leukemia of Patient PN2 (who had a previous diagnosis of nodal MCL positive for cyclin D1 by immunohistochemistry) did not show appreciable levels of cyclin D1 mRNA in this assay. The faint bands present in the lanes of Patient PN2 and one of the CLL samples are consistent with very-low-level expression from either tumor cells or “bystander” cells (e.g., normal B-lymphocytes).

Cyclin D1 mRNA overexpression measured by RT-PCR assay. Patient cases PN 1,5 and 6 showing clear cyclin D1 overexpression by reverse transcriptase–polymerase chain reaction (RT-PCR) analysis, as compared with the positive control (classical mantle cell lymphoma [MCL]). Leukemias of PN1 and PN5 were remarkable for the presence of the t(11;14) and BCL-1 gene rearrangement, respectively. PN2 had a history of classical nodal MCL; however, the subsequent blastic leukemia sample depicted here shows paradoxical loss of cyclin D1 mRNA overexpression. RNA integrity was confirmed in this case by successful amplification of β2-microglobulin mRNA (not shown). Other lanes: CLL, cases of classic chronic lymphocytic leukemia; ALL, case of adult acute lymphoblastic leukemia; −, negative control (no RNA). The faint bands in one of the CLL lanes and in lane PN2 represent very-low-level transcript products, derived from either tumor cells or background cells (e.g., residual normal B-lymphocytes) (40), detected by the sensitivity of the PCR amplification.
DISCUSSION
This report documents a distinct group of CD5-positive B-cell lymphoid leukemias that arise in elderly patients, characterized by moderate to marked leukocytosis and nonindolent behavior. The clinical presentation and cytologic features in blood and BM prompted the initial consideration of acute leukemia in the majority of these cases. However, the SIg, CD5-positive B-cell phenotype, and the absence of neutropenia are features consistent with the diagnosis of an aggressive mature B-cell lymphoid malignancy, such as transformation of CLL, B-PLL, or blastic leukemic MCL. The patients presented here do not conform clinically or phenotypically to a recently described series of six adult SIg-positive ALL, one of which was remarkable for CD5 antigen expression (7). The reported cases of these “true” SIg-positive ALL occurred at a substantially younger median age and were further distinguished by expression of the immature cell markers CD34 and TdT, in contrast to patient cases of the present study.
The differential diagnosis of atypical CD5-positive leukemias includes transformation of CLL and B-PLL. Patients in our series did not have a known previous diagnosis of CLL or other chronic (“low-grade”) B-cell lymphoproliferative disorders. Case reports of transformed CLL have been described in conjunction with chromosome 8(q24)/C-MYC gene abnormalities; however, these neoplasms usually resemble L3 (Burkitt’s type) ALL (8, 9, 10). Large cell (“Richter’s”) transformations of CLL typically manifest in nodal sites and are very unlikely to be markedly leukemic. One study documented the utility of CD23 antigen in differentiating aggressive lymphoid transformations of CLL/small lymphocytic lymphoma (SLL) from blastic MCL (11). In the present report, three of four cases studied for CD23 expression were negative for this marker and the remaining one was only weakly positive, thus arguing against a high-grade transformation from unrecognized, underlying CLL. B-PLL is cytologically characterized by moderately condensed chromatin with a single prominent nucleolus and the presence of relatively abundant cytoplasm. CD5 expression is variably observed in B-PLL, and a background of more typical CLL-type cells may be present. The t(11;14) abnormality has been reported to occur in a minority of B-PLL (12); however, other lymphoproliferative disorders, notably leukemic phase MCL, may morphologically overlap with B-PLL (13). The six cases in our series demonstrated circulating and marrow blastic lymphoid cells with high nuclear:cytoplasmic ratio and small or absent nucleoli, in contrast to typical prolymphocytoid morphology. Furthermore, splenomegaly, which is commonly observed in B-PLL, was present in only two of the six patients.
MCL is a clinically aggressive CD5-expressing B-cell neoplasm highly associated with the t(11;14)(q13;q32) abnormality and rearrangement of the BCL-1 locus, resulting in deregulated cyclin D1 expression (14, 15, 16, 17, 18, 19, 20, 21). Although the features of blastic or large cell MCL (22, 23, 24) and peripheralizing MCL (13, 25, 26, 27, 28) have each been well described in the literature, cases of blastic MCL presenting with predominant blood and marrow involvement have not been clearly identified to our knowledge. Two previous reports describe primarily leukemic disorders with some similarities to the current patient series presented here. Cuneo et al. (29) identified seven patients with t(11;14)-positive “atypical CLL” among a consecutive group of 72 CLL cases. These neoplasms were found to differ from typical CLL cases by virtue of variant morphology, karyotypic progression during disease course, bright SIg expression, and lack of CD23. Daniel et al. (30) studied five patients with t(11;14)-positive leukemic non-Hodgkin’s lymphoma with similar phenotypic features and limited or absent nodal involvement. Notable in the latter report is that three of four cases demonstrated BCL-1 or cyclin D1 gene rearrangements; however, only one case involved the MTC locus. Significant, however, is that cases with blastic cytology were not identified in the two series.
Singleton et al. (31) recently documented six cases of leukemic blastic MCL complicating preexistent nodal disease, with similar patient demographics and hematologic findings to the present report. However, the median survival in the series of Singleton et al. is considerably longer than in our patient group, possibly related to differences in therapeutic management and patient performance status. Moreover, all patients in the series of Singleton et al. had a preceding diagnosis of MCL by lymph node biopsy; this is in contrast to the dramatic initial leukemic manifestations encountered in five of the six patients in the present description. In our patient group, one case represents a blastic transformation from underlying (preexistent) MCL, though interestingly, this patient’s disease was previously documented not to involve BM or PB. Surprising is that only weak background levels of cyclin D1 mRNA were detectable by RT-PCR in the blastic tumor sample of this latter case, which may reflect a paradoxical loss of expression during transformation.
Involvement of the 8(q24)/C-MYC locus has also been reported in rare cases of transformed MCL (32; 33; Au WY et al., unpublished data), associated in some with blastic morphologic features; however, in each instance, significant concurrent nodal disease or prior classical-type nodal MCL histology has been present. For five individuals in this series, no evidence of previous extramedullary disease was elicited and 8(q24) abnormalities were not observed in two tumors with evaluable karyotypic studies. Nonetheless, variable involvement of nodal sites was evident in some members of this patient group at the time of diagnosis, indicating a lymphomatous origin or component in these cases. Important is that the blood and BM samples obtained were generally sufficient for thorough diagnostic evaluation in our series, even though lymph node or tissue biopsies likely would have provided correlative information for patients with documented extramedullary disease. Accordingly, the aggregate clinical and morphologic findings, along with the phenotypic and genetic data detailed in this study, are consistent with unusual de novo or predominant leukemic presentations of blastic MCL. In particular, among four patient tumors, the detection of the t(11;14)(q13;q32) anomaly, BCL-1 (MTC locus) gene abnormalities, and cyclin D1 mRNA overexpression strongly support this diagnosis.
Delineating this group of patients more precisely, when blood or BM examination represents the initial diagnostic procedure, requires heightened awareness and the application of ancillary immunologic and molecular techniques. The presence of BCL-1 gene rearrangements or cyclin D1 abnormalities should be assessed in such cases to establish the diagnosis of blastic mantle cell leukemia. Fluorescence in situ hybridization at the 11(q13) locus using BCL-1 flanking probes has been described as a rapid and highly sensitive means of detecting BCL-1 rearrangements in metaphase or interphase nuclei (34, 35, 36), and use of this method would obviate the relatively low detection rate of SB and DNA PCR analyses. Application of cyclin D1 immunoperoxidase has been reported to be a useful and specific diagnostic adjunct in MCL (21, 37, 38, 39, 40, 41, 42, 43); however, in our experience, this method can be inconsistent, particularly in B5-fixed and decalcified tissues. Detection of cyclin D1 mRNA overexpression by slot blotting method (44) or RT-PCR technique as illustrated here and by others (45) may provide rapid, clinically useful alternatives. To this end, the determination of increased cyclin D1 levels by Western blot or molecular methods has been shown to identify prognostically relevant patient groups among some chronic B-cell leukemias, despite the difficulty in accurately classifying many of these neoplasms by cytomorphologic criteria (46).
In summary, we describe an uncommon group of CD5-positive, immunophenotypically mature B-lymphoid tumors characterized by leukemic presentation, blastic blood and BM features, and aggressive clinical behavior. The clinical and pathologic features, in conjunction with BCL-1/cyclin D1 abnormalities, best define these neoplasms as leukemic manifestations of blastic MCL. Additional studies are required to explore the pathophysiologic basis for the marked, predominant leukemic dissemination in such cases.
References
Hammami A, Chan WC, Michels SD, Nassar VH . Mature B cell acute leukemia: a clinical, morphological, immunological and cytogenetic study of nine cases. Hematol Pathol 1991; 5: 109–118.
Molot RJ, Meeker TC, Wittwer CT, Perkins SL, Segal GH, Masih AS, et al. Antigen expression and polymerase chain reaction amplification of mantle cell lymphomas. Blood 1994; 83: 1626–1631.
Segal GH, Masih AS, Canavan Fox A, Jorgensen T, Scott M, Braylan RC . CD5-expressing B cell non-Hodgkin’s lymphomas with bcl-1 gene rearrangement have a relatively homo-geneous immunophenotype and are associated with an overall poor prognosis. Blood 1995; 85: 1570–1579.
Williams ME, Meeker TC, Swerdlow SH . Rearrangement of the chromosome 11 bcl-1 locus in centrocytic lymphoma: analysis with multiple breakpoint probes. Blood 1991; 78: 493–498.
Williams ME, Swerdlow SH, Rosenberg CL, Arnold A . Chromosome 11 translocation breakpoints at the Prad1/cyclin d1 gene locus in centrocytic lymphoma. Leukemia 1993; 7: 241–245.
Pinyol M, Campo E, Nadal A, Terol JM, Jares P, Nayach Irazema ; et al. Detection of the bcl-1 rearrangement at the major translocation cluster in frozen and paraffin-embedded tissues of mantle cell lymphomas by polymerase chain reaction. Am J Clin Pathol 1996; 105: 532–537.
Vasef MA, Brynes RK, Murata-Collins JL, Arber DA, Medeiros LJ . Surface immunoglobulin light chain-positive acute lymphoblastic leukemia of FAB L1 or L2 type: a report of 6 cases in adults. Am J Clin Pathol 1998; 110: 143–149.
Asou N, Osata M, Horikawa K, Nishikawa K, Sakitani O, Li L, et al. Burkitt’s type acute lymphoblastic transformation associated with t(8;14) in a case of B cell chronic lymphocytic leukemia. Leukemia 1997; 11: 1986–1988.
Litz CE, Arthur DC, Gajl-Peczalska KJ, Rausch D, Copenhaver C, Coad JE, et al. Transformation of chronic lymphocytic leukemia to small non-cleaved cell lymphoma: a cytogenetic, immunological and molecular study. Leukemia 1991; 5: 972–978.
Mohamed AN, Compean R, Dan ME, Smith MR, Al-Katib A . Clonal evolution of chronic lymphocytic leukemia to acute lymphoblastic leukemia. Cancer Genet Cytogenet 1996; 86: 143–146.
Dunphy CH, Wheaton SE, Perkins SL . CD23 expression in transformed small lymphocytic lymphomas/chronic lymphocytic leukemias and blastic transformations of mantle cell lymphoma. Mod Pathol 1997; 10: 818–822.
Brito-Babapulle V, Ellis J, Matutes E, Oscier D, Khokhar T, MacLennan K, et al. Translocation t(11;14)(q13;q32) in chronic lymphoid disorders. Genes Chromosomes Cancer 1992; 5: 158–165.
Cohen PL, Kurtin PJ, Donovan KA, Hanson CA . Bone marrow and peripheral blood involvement in mantle cell lymphoma. Br J Haematol 1998; 101: 302–310.
Campo E, Raffeld M, Jaffe ES . Mantle cell lymphoma. Semin Hematol 1999; 36: 115–127.
Weisenburger DD, Armitage JO . Mantle cell lymphoma: an entity comes of age. Blood 1996; 87: 4483–4494.
Argatoff LH, Connors JM, Klasa RJ, Horsman DE, Gascoyne RD . Mantle cell lymphoma: a clinicopathologic study of 80 cases. Blood 1997; 89: 2067–2078.
Samaha H, Dumontet C, Ketterer N, Moullett I, Thieblemont C, Bouafia F, et al. Mantle cell lymphoma: a retrospective study of 121 cases. Leukemia 1998; 12: 1281–1287.
Rimokh R, Berger F, Delsol G, Charrin C, Bertheas MF, Ffrench M, et al. Rearrangement and overexpression of the BCL-1/PRAD-1 gene in intermediate lymphocytic lymphomas and in t(11q13)-bearing leukemias. Blood 1993; 81: 3063–3067.
Oka K, Ohno T, Kita K, Yamaguchi M, Takakura N, Nishii K, et al. PRAD1 gene overexpression in mantle cell lymphoma but not in other low grade B-cell lymphomas, including extranodal lymphoma. Br J Haematol 1994; 86: 786–791.
Bosch F, Jares P, Campo E, Lopez-Guillermo A, Piris MA, Villamor N, et al. PRAD1/cyclin d1 gene overexpression in chronic lymphoproliferative disorders: a highly specific marker of mantle cell lymphoma. Blood 1994; 84: 2726–2732.
de Boer CJ, van Krieken JM, Schuuring E, Kluin PM . Bcl-1/cyclin D1 in malignant lymphoma. Annu Oncol 1997; 8 (Suppl 2): S109–S117.
Lardelli P, Bookman MA, Sundeen J, Longo DL, Jaffe ES . Lymphocytic lymphoma of intermediate differentiation: morphologic and immunophenotypic spectrum and clinical correlations. Am J Surg Pathol 1990; 14: 752–763.
Ott MM, Ott G, Kuse R, Porowski P, Gunzer U, Feller AC, et al. The anaplastic variant of centrocytic lymphoma is marked by frequent rearrangements of the bcl-1 gene and high proliferation indices. Histopathology 1994; 24: 329–334.
Zoldan MC, Inghirami G, Masuda Y, Vandekerckhove F, Raphael B, Amorosi E, et al. Large cell variants of mantle cell lymphoma: cytologic characteristics and p53 anomalies may predict poor outcome. Br J Haematol 1996; 93: 475–486.
Pittaluga S, Verhoef G, Criel A, Criel A, Maes AM, Nuyts JM, et al. Prognostic significance of bone marrow trephine and peripheral blood smears in 55 patients with mantle cell lymphoma. Leuk Lymphoma 1996; 21: 115–125.
Shivdasani RA, Hess JL, Skarin AT, Pinkus GS . Intermediate lymphocytic lymphoma: clinical and pathologic features of a recently characterized subtype of non-Hodgkin’s lymphoma. J Clin Oncol 1993; 11: 802–811.
Pombo de Oliveira MS, Jaffe ES, Catovsky D . Leukaemic phase of mantle zone (intermediate) lymphoma: its characterisation in 11 cases. J Clin Pathol 1989; 42: 962–972.
Vadlamudi G, Lionetti KA, Greenberg S, Mehta K . Leukemic phase of mantle cell lymphoma. Two case reports and review of the literature. Arch Pathol Lab Med 1996; 120: 35–40.
Cuneo A, Balboni M, Piva N, Rigolin GM, Roberti MG, Mejak C, et al. Atypical chronic lymphocytic leukemia with t(11;14)(q13;q32): karyotype evolution and prolymphocytic transformation. Br J Haematol 1995; 90: 409–416.
Daniel MT, Tigaud I, Flexor MA, Nogueira ME, Berger R, Jonveaux P . Leukaemic non-Hodgkin’s lymphomas with hyperdiploid cells and t(11;14)(q13;q32): a subtype of mantle cell lymphoma? Br J Haematol 1995; 90: 77–84.
Singleton TP, Anderson MM, Ross CW, Schnitzer B . Leukemic phase of mantle cell lymphoma, blastoid variant. Am J Clin Pathol 1999; 111: 495–500.
Leroux D, Le Marc'Hadour F, Gressin R, Jacob MC, Keddari E, Monteil M, et al. Non-Hodgkin's lymphomas with t(11;14)(q13;q32): a subset of mantle zone/intermediate lymphocytic lymphoma? Br J Haematol 1991; 77: 346–353.
Tirier C, Zhang Y, Plendl H, Weber-Matthiesen K, Langer W, Schlegerlberger B . Simultaneous presence of t(11;14) and a variant Burkitt’s translocation in the terminal phase of a mantle cell lymphoma. Leukemia 1996; 10: 346–350.
Monteil M, Callanan M, Dascalescu C, Sotto JJ, Leroux D . Molecular diagnosis of t(11;14) in mantle cell lymphoma using two-colour interphase fluorescence in situ hybridization. Br J Haematol 1996; 93: 656–660.
Coignet LJA, Schuuring E, Kibbelaar RE, Raap TK, Kleiverda KK, Bertheas MF, et al. Detection of 11q13 rearrangements in hematologic neoplasias by double-colour fluorescence in situ hybridization. Blood 1996; 87: 1512–1519.
Vaandrager JW, Shuuring E, Zwikstra E, de Boer CJ, Kleiverda KK, Han J, et al. Direct visualization of dispersed 11q13 chromosomal translocations in mantle cell lymphoma by multicolor DNA fiber fluorescence in situ hybridization. Blood 1996; 88: 1177–1182.
Yang WI, Zukerberg LR, Motokura T, Arnold A, Harris NL . Cyclin D 1 (Bcl-1, PRAD1) protein expression in low-grade B-cell lymphomas and reactive hyperplasia. Am J Pathol 1994; 145: 86–96.
Zukerberg LR, Yang WI, Arnold A, Harris NL . Cyclin D 1 expression in non-Hodgkin’s lymphomas: detection by immunohistochemistry. Am J Clin Pathol 1995; 103: 756–760.
de Boer CJ, Schuuring E, Dreef E, Peters G, Bartek J, Kluin PM, et al. Cyclin D 1 protein analysis in the diagnosis of mantle cell lymphoma. Blood 1995; 86: 2715–2723.
Alkan S, Schnitzer B, Thompson JL, Moscinski LC, Ross CW . Cyclin D 1 protein expression in mantle cell lymphoma. Ann Oncol 1995; 6: 567–570.
Ott MM, Bartkova J, Bartek J, Durr A, Fischer L, Ott G, et al. Cyclin D 1 expression in mantle cell lymphoma is accompanied by downregulation of cyclin D3 and is not related to proliferative activity. Blood 1997; 90: 3154–3159.
Vasef MA, Medeiros LJ, Koo C, McCourty A, Brynes RK . Cyclin D 1 Immunohistochemical staining is useful in distinguishing mantle cell lymphoma from other low-grade B-cell neoplasms in bone marrow. Am J Clin Pathol 1997; 108: 302–307.
Soslow RA, Zukerberg LR, Harris NL, Warnke RA . BCL-1 (PRAD1/cyclin D1) overexpression distinguishes the blastoid variant of mantle cell lymphoma from B-lineage lymphoblastic lymphoma. Mod Pathol 1997; 10: 810–817.
Mauvieux L, Canioni D, Hermine O, Valensi F, Radford-Weiss I, Azagury M, et al. Quantitative RNA slot-blot analysis of CCND1/cyclin D1 expression in suspected mantle cell lymphoma. Leukemia 1998; 12: 78–85.
Uchimaru K, Taniguchi T, Yoshikawa M, Asano S, Arnold A, Fujita T, et al. Detection of cyclin D1 (bcl-1, PRAD1) overexpression by a simple competitive reverse transcription-polymerase chain reaction assay in t(11;14)(q13;q32)-bearing B cell malignancies and/or mantle cell lymphoma. Blood 1997; 89: 965–974.
Levy V, Ugo V, Delmer A, Tang R, Ramond S, Perrot JY, et al. Cyclin D1 overexpression allows identification of an aggressive subset of leukemic lymphoproliferative disorder. Leukemia 1999; 13: 1343–1351.
Acknowledgements
The authors gratefully acknowledge Drs. H. Mooney, S. Mastovich, C. Sever, and H. Johnson for providing case material and patient information. In addition, we thank the members of the UNM Cancer Center and BC Cancer Agency Molecular Diagnostics laboratories for excellent technical assistance and Ms. J. Lusk for help in manuscript preparation.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Viswanatha, D., Foucar, K., Berry, B. et al. Blastic Mantle Cell Leukemia: An Unusual Presentation of Blastic Mantle Cell Lymphoma. Mod Pathol 13, 825–833 (2000). https://doi.org/10.1038/modpathol.3880144
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880144
Keywords
This article is cited by
-
Non-Hodgkin lymphoma mimicking acute leukemia: a report of six cases and review of the literature
Journal of Hematopathology (2022)
-
Palatine Tonsils Primary Presentation of Blastoid Variant of Mantle Cell Lymphoma: Case Report
Head and Neck Pathology (2021)
-
Evaluation of peripheral blood involvement of mantle cell lymphoma by fluorescence in situ hybridization in comparison with immunophenotypic and morphologic findings
Modern Pathology (2004)
-
Leukemic Mantle Cell Lymphoma: Clinical and Pathologic Spectrum of Twenty-Three Cases
Modern Pathology (2001)
