
Abstract
Pagetoid reticulosis (PR), also known as Woringer-Kolopp disease, is a form of cutaneous T-cell lymphoma that demonstrates striking epidermotropism on histologic examination. We present the histologic, immunologic, and molecular findings for seven patients who had PR. The patients ranged in age from 33 to 67 years. All patients presented with one or several thick plaques involving the distal extremities except for one patient, who presented with a tongue lesion. Immunohistochemical staining of the atypical lymphoid cells demonstrated a T-cell phenotype in all cases. In one of four frozen cases, the neoplastic cells were of T-helper cell phenotype (CD4 positive). Four of seven cases demonstrated a T-cytotoxic/suppressor cell phenotype (CD8 positive). The T-cell subset for the remaining two cases could not be determined. CD30 positivity and a high growth fraction as indicated by staining with Ki-67 were seen in three of seven and three of four cases, respectively. Genotypic analysis performed on three of our cases revealed T-cell receptor (γ and/or β) rearrangement, indicating a clonal proliferation. The clinical follow-up ranged from 15 months to 13 years. Four of seven patients are alive and free of disease after treatment with excision or local irradiation. One patient relapsed twice after treatment with radiation and photochemotherapy with 8-methoxypsoralen and UVA and was then lost to follow-up. The lesions of another patient resolved spontaneously but recurred at the same and in an additional site 5 years later. One patient recurred after electron beam therapy. The recurrent lesion improved with radiation therapy and local wound care but never resolved completely. The patient died of unrelated causes. Our findings suggest that PR is a distinct clinicopathologic entity, separate from unilesional mycosis fungoides, demonstrating a slow disease course. The disease is a clonal cutaneous T-cell lymphoma with relatively consistent clinical and histopathologic findings but a heterogeneous immunophenotypic profile.
Similar content being viewed by others


INTRODUCTION
The term pagetoid reticulosis (PR) was introduced in 1973 by Braun-Falco et al. (1) to characterize a rare skin disorder, originally described by Woringer and Kolopp in 1939 (2). The term was used to acknowledge the similarity of the epidermotropic atypical cells to the intraepidermal adenocarcinomatous cells found in Paget’s disease of the nipple. Two variants of the disease are described: the localized type (Woringer-Kolopp disease [WKD]) and the disseminated type (Ketron-Goodman disease [KGD]). WKD typically presents as a solitary, slowly growing cutaneous plaque on the extremities. The lesional epidermis is markedly acanthotic and is infiltrated by cytologically atypical mononuclear cells demonstrating striking epidermotropism. The histologic features of KGD are identical to those of WKD. The distinction between the two entities rests on clinical grounds alone. The Ketron-Goodmann variant is characterized by dissemination of lesions, possible association with other lesions characteristic of mycosis fungoides (MF), and, at times, progression to fatal outcome. Some authors have suggested that because of its more aggressive biologic behavior, KGD should be regarded as a variant of MF showing prominent epidermotropism and that the term PR should be reserved for the indolent form of the disease (WKD) (3). The nature of the atypical cells in PR was the subject of much controversy in literature over the past years. At present, however, immunophenotypic and genophenotypic studies have clearly established a T-cell origin and monoclonality of the atypical epidermotropic cells. Cases of PR with T-helper phenotype (CD4), T-cytotoxic/suppressor (CD8) phenotype, and CD4/CD8 double-negative phenotype are described (4, 5, 6).
Most of the literature regarding PR consists of small series with only a few patients and short clinical follow-up. Moreover, most immunophenotypic studies included only a limited antibody panel. Our study includes seven patients with long-term follow-up, up to 13 years. In addition, each case was studied with an extensive antibody panel.
MATERIALS AND METHODS
Case Selection
The cases were obtained from the files of Stanford University Hospital, University of California at San Francisco Medical Center, and the Department of Dermatology of the Ludwig-Maximilians-University, Munich, Germany. Histologic sections and immunohistochemical studies performed at the time of diagnoses were reviewed, and the diagnoses were confirmed.
Immunohistochemical Studies
Immunohistochemical studies were performed on paraffin and/or frozen tissue sections using a biotin-streptavidin method previously described (7). Primary antibodies were directed against CD2, CD3, CD4, CD5, CD7, CD8, CD20, CD25, CD45, CD45RA, CD45RB, CD45RO, TCRβ, TIA-1, CD30, CD56, and Ki-67 (summarized in Table 1). For paraffin-section immunohistochemistry, serial 5-μm-thick paraffin sections were deparaffinized in xylene, and endogenous peroxidase was blocked by incubation with 3% H2O2 for 8 min. For frozen-section immunohistochemistry, cryostat sections were fixed in acetone at 4° C for 10 min.
T-Cell Rearrangement Studies
T-cell receptor (TCR) gene rearrangement studies were performed in three cases. The amount of tissue remaining after histologic examination and immunohistochemical studies in the remaining four cases was insufficient for molecular analysis, even by polymerase chain reaction (PCR). Two of the cases (Cases 3 and 4) were previously published as case reports and genotypic analyses of the TCR β chain (Case 4), and the TCR β and γ chains (Case 3) were performed during the initial workup (8, 9). In Case 7, the TCR γ chain was analyzed by PCR and polyacrylamide gel electrophoresis, as described previously (10).
RESULTS
Clinical Data
Our patients included five women and two men, ranging in age from 33 to 67 years. Four patients presented with one or several, often confluent, thick, erythematous, scaly plaques on the forearms or hands; two of the patients had plaques on their lower legs or feet (Fig. 1), and one presented with a papule on the tongue. Specific sites of involvement and approximate size of affected areas are listed in Table 2. Duration of the lesions before diagnosis was known in all but one case and ranged from 3 months to 40 years. The lesions ranged in size from 0.6 cm to more than 30 cm. When available, the clinical history indicated that the disease was asymptomatic, except for Case 3, the largest tumor in this series, covering the hand and most of the forearm. The affected area was superinfected and ulcerated; at one point, the ulceration extended to the bone and consideration was given to amputation of the extremity. Treatment modalities included surgical excision, local radiation therapy, electron beam therapy, photochemotherapy with 8-methoxypsoralen and UVA (PUVA) baths, and local wound care (Table 2). In one case (Case 2), the disease was not treated. Follow-up information was available in all cases, with follow-up periods ranging from 15 months to 11 years. Three of our patients experienced recurrence. The recurrences involved the same and different sites (see Table 2). The patient who experienced spontaneous resolution of the initial plaques (Case 2) experienced a recurrence at the same site (hand) and also developed a lesion on the foot 5 years after spontaneous resolution. One patient (Case 7) relapsed twice. The first relapse followed complete resolution with radiation therapy; the second relapse occurred after achieving complete remission with PUVA baths. The second relapse was again treated with radiation therapy, and the patient was then lost to follow-up. Patient 3’s tumor initially responded to radiation therapy but recurred at the same site 2 years later. The recurrent lesion was large but eventually was controlled with radiation therapy and aggressive wound care; however, the lesion never resolved, and the patient died 8 years later of a cerebrovascular accident. The other six patients are alive. In one patient (Case 4), a foot lesion that was diagnosed as WKD resolved, but a separate eruption that was clinically and histologically distinct from the foot lesion developed later on the right buttock and was diagnosed as MF. The latter was treated, and the patient is free of disease 13 years after diagnosis of PR of the foot and 7 years after diagnosis of MF on the buttock. The clinical presentation of Case 5 as a tongue lesion is distinctly different from the other cases, and PR in this location has not previously been described. The patient was a 43-year-old woman who presented with a papule of unknown duration on the dorsal aspect of the tongue. The lesion was excised and diagnosed as PR. Further workup revealed no involvement of other sites. Three months after initial presentation, the patient developed an erythematous papule on the back, surrounded by a wheal (total diameter, 1.5 cm). The clinical impression was that of an insect bite. The papule was biopsied, and histology revealed a superficial and deep, dense, angiocentric lymphoid infiltrate, suspicious for angiocentric T-cell lymphoma with an exuberant arthropod bite in the differential diagnosis. Immunophenotyping revealed a T-cell phenotype without expression of the natural killer cell marker CD56. The lesion resolved but recurred 2 months later. At that point, the clinical differential diagnosis included herpes simplex virus infection, impetigo, and less likely an infiltrative process. The histology was again suspicious for an angiocentric T-cell lymphoma, consisting of a deep, dense, and angiocentric infiltrate with cytologic atypia. PCR for TCR rearrangement did not show a clonal proliferation. A systemic workup was negative for other sites of involvement, and the patient was treated with local radiation therapy. She is without evidence of disease 2 years later. In retrospect, both lesions on the back probably represented an exuberant arthropod bite reaction.

Clinical appearance of pagetoid reticulosis. Case 4: The dorsal and posterior aspects of the foot are involved by large hyperkeratotic and focally ulcerated plaques.
Histology
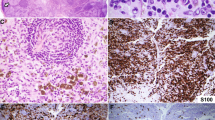
All cases revealed similar histopathologic findings (Fig. 2). The epidermis (the mucosa in Case 5) was markedly acanthotic and infiltrated by atypical mononuclear cells with a moderate amount of lightly eosinophilic cytoplasm. The cells were larger than mature lymphoid cells and contained a large nucleus with irregular borders. The nucleus was surrounded by a clear space (halo) that replaced most of the cytoplasm (Fig. 2C). A much less striking dermal inflammatory infiltrate was also present. The dermal infiltrate was composed predominantly of mature and reactive lymphocytes, histiocytes, and a few plasma cells. Eosinophils were not identified in any of the biopsies. Atypical mononuclear cells either were absent or were present only in small numbers in the dermal infiltrate.

Histopathologic appearance of pagetoid reticulosis. The epidermis is markedly acanthotic and covered by hyperkeratosis and parakeratosis (A). Particularly, the lower half of the epidermis is infiltrated by atypical lymphoid cells with enlarged, hyperchromatic and hyperconvoluted nuclei; irregular nuclear outlines; and abundant, clear cytoplasm (B and C).
Immunophenotypic Findings
The results of immunophenotypic stains were scored separately for the intraepidermal atypical cells and the underlying dermal infiltrate. The dermal infiltrate seemed to consist mainly of reactive T-cells, as evidenced by a predominance of CD3-positive, CD8-positive cells. In addition, there were scattered CD30-positive, CD56-positive, and Ki-67–positive cells. The remainder of the immunophenotypic results pertain to the atypical intraepidermal cells (Table 3). Immunohistochemical analysis of the atypical epidermotropic mononuclear cells demonstrated a T-cell phenotype in all cases (CD2 positive, CD3 positive, and CD5 positive). Antibodies against CD2 and CD5 were applied only to the four cases for which frozen tissue was available. CD4 staining was also performed only on cases for which frozen tissue was available and showed reactivity in one of four cases. Antibodies against CD8 were applied to both paraffin and frozen tissue; lesional cells were positive in four of seven cases. Three of four cases with T-cytotoxic/suppressor phenotype (CD8 positive) seemed to be composed of memory T-cells (CD45RO positive), expressed the α/β TCR on the cell surface (BF1 positive), and had a cytotoxic phenotype, as evidenced by reactivity with TIA-1. Antibodies against CD7 were applied to frozen tissue only, and one of four cases revealed staining of between 25% and 50% of epidermal cells. In the three remaining cases, the pan T-cell marker CD7 exhibited no reactivity with intraepidermal tumor cells. The B-cell marker CD20 was negative in all six cases that were tested. The intraepidermal cells did not express CD45RA in the three evaluable cases. Antibodies against CD45RB/4KB5 recognized nearly all lesional cells in two cases, were nonreactive in three cases, and stained fewer than 25% of intraepidermal cells in one case. None of the five cases that were tested for the natural killer cell marker CD56 showed convincing reactivity. Four cases were negative, and one case could not be interpreted secondary to high background staining. In three of seven cases, more than 50% of intraepidermal cells stained for CD30; the remaining cases were nonreactive. The proliferation marker Ki-67 demonstrated a proliferation rate in excess of 50% in three of four cases and labeled fewer than 5% of nuclei in the remaining case.
Genotyping
Genotypic analysis was performed on Cases 3, 4, and 7. Case 3 showed rearrangement of the TCR β and γ chains by Southern blotting. Case 4 was tested only for rearrangement of the TCR β chain by Southern blotting and showed a clonal band. Case 7 was clonal by PCR of the TCR γ chain followed by polyacrylamide gel electrophoresis.
DISCUSSION
WKD, or localized PR, was originally described 60 years ago (2). Many aspects of the disease are still the subject of controversy. These include basic issues, such as whether localized PR is an inflammatory or neoplastic disorder; if neoplastic, whether it is benign or malignant; and the relationship of PR to MF. The early literature concentrates mainly on the nature of the pagetoid intraepidermal cells that are the histologic hallmark of the disease. Some authors speculate that the atypical cells are related to Merkel cells (11). Others favor a histiocytic origin based on ultrastructural and enzyme histochemical observations (12, 13). However, more recent studies unequivocally establish a T-cell lineage for the atypical epidermotropic cells (4, 8, 9, 14, 15, 16, 17). Further subtyping reveals three different phenotypes for the large epidermotropic cells, namely a CD4-positive T-helper phenotype (4, 17, 18, 19), CD8-positive T-cytotoxic/suppressor phenotype (4, 5, 6, 9), or a CD4/CD8 double-negative phenotype (6, 8, 14, 20). The results of the current study are in keeping with the observations described in the literature. In all seven cases, the neoplastic cell population consisted of T cells (CD45 positive, CD2 positive, CD3 positive, CD5 positive, CD20 negative). In four of seven cases, the epidermotropic cells comprised CD8-positive T-cytotoxic/suppressor cells. One case demonstrated a CD4-positive T-helper cell phenotype, and two cases could not be further subtyped. For both cases, we had only paraffin-embedded but no frozen material available. Both cases did not react with the CD8 antibody in paraffin. These two cases could be of either CD4-positive T-helper cell or CD4/CD8 double-negative phenotype. We also confirmed the observation of other investigators (6, 9, 18) that CD7 expression is often either aberrantly decreased or completely absent in cases of PR, whereas CD2, CD3, and CD5 expression is preserved (see Table 3). CD7 expression is markedly decreased or lost in a high percentage of cutaneous T-cell lymphomas (21), but this is not specific for T-cell malignancies and can also be observed in inflammatory dermatoses (22).
Several previous studies examined CD45RO (UCHL-1) expression in localized PR, a marker for memory T cells. At least nine reported cases demonstrated lack of CD45RO staining (6, 20, 23). Conversely, at least four cases tested positive for CD45RO (18, 19). In a more detailed analysis of the CD45 complex, Sterry and Hauschild (20) examined two cases of localized PR and found complete loss of the CD45 complex on the cell surface, as evidenced by lack of staining for CD45 (leukocyte common antigen), CD45RO (UCHL1), CD45RA (Leu18), and CD45RB (4KB5). Similar results were obtained by Mielke et al. (6), who examined two cases of PR and in addition to lack of staining for CD45RO found absent staining for CD45R (2H4) and staining of only a subset of cells for CD45RB (4KB5). Lack of expression of CD45RA and CD45RB may be seen in peripheral T cells; however, lack of staining for CD45 and CD45RO is unusual for most cutaneous lymphomas of T-cell origin. Sterry and Hauschild (20) therefore suggested that lack of CD45, CD45RO, and CD45RA expression may be helpful in distinguishing localized PR from other cutaneous T-cell lymphomas. Our results with all three cases that stained for CD45 (LCA) and four of five cases that stained for CD45RO question the utility of these antibodies in the differential diagnosis of PR. Our results are more in keeping with the findings of Burns et al. (18), who also detected CD45RO and CD45R reactivity in all three of their cases. It is unlikely that the choice of antibody for the CD45RO epitope contributed to the different results, as A6 and UCHL-1 show a very comparable staining profile (24).
Alaibac et al. (14, 25) reported a case of PR that was CD4/CD8 double negative and expressed the γ/δ TCR on the cell surface. The authors speculated that several of the CD4/CD8 double-negative cases that were reported in the literature may represent γ/δ T-cell lymphomas, although other studies had not tested for this possibility. Our study did not add any definite cases with a similar phenotype.
Most authors regard PR as a cutaneous lymphoproliferative disorder with indolent clinical behavior (8, 9, 14, 20). In its prototypical form, the disease presents as scaling patches on the distal extremities that over years slowly progress to plaques and nodules. The disease usually responds to excision or local radiotherapy. It can recur but has little propensity for dissemination or visceral involvement. A neoplastic nature is supported by several studies that showed clonal T-cell receptor rearrangements by Southern blotting (8, 9, 23). Mielke et al. (6) chose the term benign cutaneous lymphoma to convey the same concept of a clonal lymphoid proliferation with an indolent clinical course for PR. Burns et al. (18) shared Mielke’s view and nomenclature for PR. Crowson and Magro (19) took the view that PR is a lymphomatoid hypersensitivity reaction to an arthropod assault. They described two patients who presented with small plaques on the extremities after receiving spider bites. Both lesions were excised and did not recur. Neither case showed clonal TCR rearrangement by Southern blotting. The clinical course and molecular analysis led the authors to conclude that PR is an entity that is distinct from MF and that is reactive rather than neoplastic. It is of note, however, that the plaques in both cases measured only 1.0 and 1.2 cm, respectively, and were considerably smaller than most lesions of PR. Furthermore, the histologic features of Case 2 were atypical for PR. The picture shows a predominantly dermal infiltrate with only minimal epidermotropism, instead of the almost exclusively epidermal distribution of the neoplastic cells in PR. On the basis of the clinical course of our cases and careful review of the literature, we propose a more cautious approach. In our opinion, PR is a cutaneous T-cell lymphoma with uncertain clinical behavior. Each of the three cases in our series that could be evaluated with molecular methods showed clonal rearrangement of the TCR. In terms of clinical course, three patients (Cases 2, 3, and 7) experienced one or several recurrences. Patient 3 never achieved a complete remission despite local debridement, local wound care, and x-ray therapy. At one point during her course, amputation of her forearm had to be considered because the fungating tumor was a continuous source of infection. One patient (Case 4) developed Stage Ia MF on the buttock several years after the lesions of PR completely resolved after local radiation therapy. Another patient presented with a tongue lesion (Case 5), and although she did not experience a local recurrence, she developed on the back an atypical angiocentric lymphoid infiltrate that could not be diagnosed unequivocally. Differential diagnostic considerations of the lesion on the back were between an angiocentric T-cell lymphoma and an exuberant arthropod bite reaction. Two patients (Cases 1 and 6) demonstrated a more typical course of complete remission after local excision and radiotherapy, respectively, but the follow-up for both of these patients is relatively short: 15 months and 2 years, respectively. It seems that the clinical course of PR is less predictable than generally thought, and long-term clinical follow-up is warranted. A review of the literature reveals more cases that did not follow the prototypical course and showed recurrence or progression. Ioannides et al. (26) reported a case of PR with a plaque on the foot that continued to enlarge over a period of several years despite treatment with topical and intralesional steroids, x-ray therapy, and salicylic acid. After excision with clear margins, the tumor recurred in the same area. Yagi et al. (23) described a 54-year-old woman who had PR and who after 2 years progressed to disseminated disease. A 68-year-old man described by Tan et al. (27) experienced numerous recurrences after several treatments with radiotherapy and 5-fluorouracil cream. Given the small number of reported cases of PR, our patients in conjunction with the cases described above represent a significant percentage in which the clinical course deviated from the alleged indolent behavior of PR. In a very thoroughly researched and thoughtful editorial, Zackheim (28) advocated a similar view of PR as a relatively indolent cutaneous lymphoma with potential for frankly malignant behavior, warranting aggressive therapy.
Another controversial aspect of PR is its relationship to MF (5, 16, 18, 27, 28, 29, 30, 31, 32). Much of the controversy arises from the clinical and microscopic similarities between the two entities, the lack of established diagnostic criteria for PR, and, as a result, different interpretations by different authors. Nevertheless, there are several clinical, histologic, and immunophenotypic features that distinguish PR from MF (see Table 4). In PR, the atypical lymphoid cells are almost exclusively confined to the epidermis and sometimes have a histiocyte-like morphology. In contrast, MF is characterized by cells with hyperchromatic and hyperconvoluted nuclei that are present both above and below the dermal-epidermal junction. The distribution between the two compartments does not seem to change even in well-advanced and thick lesions. In PR, the lesional thickness can be accounted for by marked acanthosis accompanied by prominent epidermotropism, whereas plaques and tumors in MF are caused by dermal infiltration and expansion by tumor cells. In addition, the dermal inflammatory infiltrate in PR is composed primarily of reactive lymphocytes and histiocytes. Eosinophils are characteristically absent in this disease (33; personal observations). Conversely, dermal infiltrates in late patches and plaques of MF are usually more polymorphous and frequently contain eosinophils (34). Clinically, there is little resemblance between the classic form of MF and PR. MF typically involves the trunk, buttocks, and proximal extremities, with several or many lesions, and plaques and tumors seldom are verrucous. Solitary PR typically affects the distal extremities, and advanced lesions can be verrucous and hyperkeratotic. The controversy is mainly centered on the distinction of mycosis palmaris et plantaris (MFPP) from PR. MFPP is a rare manifestation of MF (0.6% of cases [29]) that clinically presents as thin acral plaques that mimic chronic hand or foot dermatitis. Several cases of MFPP showed recurrence or progression (29, 31). It seems, however, that progression in MFPP consists of spread to other acral sites without accompanying thickening of the plaques or formation of tumors. This is in contrast to PR, which shows a distinct propensity to form thick plaques and tumors. Finally, there are several differences in immunophenotype between MF and PR. In MF, the tumor cells are CD4-positive T-helper cells. In contrast, the neoplastic cells in PR may be CD4 positive, CD8 positive (T-cytotoxic/suppressor), or CD4/CD8 double negative. The proliferation rate, as measured by staining with the monoclonal antibody Ki-67, is usually below 10% in patch and plaque lesions of MF (35). In contrast, several studies, including ours (see Table 3), demonstrated in excess of 30% Ki-67–positive cells in PR (6, 8, 9, 36, 37). Similarly, MF only rarely contains CD30 (Ki-1)-positive cells (38, 39), whereas CD30 reactivity is variable in PR. Occasional cases showed a high percentage (>50%) of CD30-positive tumor cells (6, 9), but other studies that tested for CD30 in PR found no expression (8, 36). In our study, three of seven cases of PR were strongly positive for CD30 (>50% of cells).
In summary, our findings suggest that PR is a distinct clinicopathologic entity, separate from MF, with relatively uniform clinical and histologic findings but a heterogeneous immunophenotypic profile. It seems that the clinical behavior is less predictable than initially thought and that aggressive local management and close long-term follow-up are warranted. Solitary PR should always be considered in the differential diagnosis of strikingly epidermotropic forms of cutaneous T-cell lymphoma. We believe that if careful attention is given to the clinical, histologic, and phenotypic findings, the differentiation between PR and MFPP can be made.
References
Braun-Falco O, Marghescu S, Wolf HH . Pagetoid reticulose (Morbus Woringer-Kolopp). Hautarzt 1973; 24: 11–21.
Woringer FR, Kolopp P . Lésion érythémato-squameuse polycyclique de l’avant-bras évoluant depuis 6 ans chez un garçonnet de 13 ans. Ann Dermatol Venereol 1939; 10: 945–948.
Lacour JP, Juhlin L, el Base P, Barety M, Ortonne JP . Disseminated pagetoid reticulosis associated with mycosis fungoides: immunomorphologic study. J Am Acad Dermatol 1986; 14: 898–901.
Deneau DG, Wood GS, Beckstead J, Hoppe RT, Price N . Woringer-Kolopp disease (pagetoid reticulosis). Four cases with histopathologic, ultrastructural, and immunohistologic observations. Arch Dermatol 1984; 120 (8): 1045–1051.
Mackie RM, Turbitt ML . A case of pagetoid reticulosis bearing the T cytotoxic suppressor surface marker on the lymphoid infiltrate: further evidence that pagetoid reticulosis is not a variant of mycosis fungoides. Br J Dermatol 1984; 110 (1): 89–94.
Mielke V, Wolff HH, Winzer M, Sterry W . Localized and disseminated pagetoid reticulosis. Diagnostic immunophenotypical findings. Arch Dermatol 1989; 125 (3): 402–406.
Bindl JM, Warnke RA . Advantages of detecting monoclonal antibody binding to tissue sections with biotin and avidin reagents in Coplin jars. Am J Clin Pathol 1986; 85: 490–493.
Wood GS, Weiss LM, Hu CH, Abel EA, Hoppe RT, Warnke RA, et al. T-cell antigen deficiencies and clonal rearrangements of T-cell receptor genes in pagetoid reticulosis (Woringer-Kolopp disease). N Engl J Med 1988; 318 (3): 164–167.
Smoller BR, Stewart M, Warnke R . A case of Woringer-Kolopp disease with Ki-1 (CD30) + cytotoxic/suppressor cells. Arch Dermatol 1992; 128 (4): 526–529.
Toro JR, Sander CA, LeBoit PE . Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? A study by light microscopy and molecular methods. Am J Dermatopathol 1997; 19: 108–118.
Revuz J, Pouget F, Allegret C, Wechsler J, Jouffroy L, Touraine R . La réticulose pagétoïde de Woringer-Kolopp: une maladie de la cellule de Merkel. Ann Dermatol Venereol 1977; 104: 312–320.
Kerdel FA, MacDonald DM . Pagetoid reticulosis. Histiocyte marker studies. Arch Dermatol 1984; 120 (1): 76–79.
Chu AC, MacDonald DM . Pagetoid reticulosis: a disease of histiocytic origin. Br J Dermatol 1980; 103 (2): 147–157.
Alaibac M, Chu AC . Woringer-Kolopp disease. Am J Surg Pathol 1996; 20 (9): 1153–1154.
Gonzalez M, Martin PM, San MJ, Caballero MD, Lopez BA . Phenotypic characterization of skin-infiltrating cells in pagetoid reticulosis by monoclonal antibodies. Acta Dermatol Venereol 1984; 64 (5): 421–424.
Jones RR, Chu A . Pagetoid reticulosis and solitary mycosis fungoides. Distinct clinicopathological entities. J Cutan Pathol 1981; 8 (1): 40–51.
Slater D, Goepel J, Walker A, Corbett P . Lymphocyte subsets in pagetoid reticulosis. Br J Dermatol 1984; 111 (2): 244–246.
Burns MK, Chan LS, Cooper KD . Woringer-Kolopp disease (localized pagetoid reticulosis) or unilesional mycosis fungoides? An analysis of eight cases with benign disease. Arch Dermatol 1995; 131 (3): 325–329.
Crowson AN, Magro CM . Woringer-Kolopp disease. A lymphomatoid hypersensitivity reaction. Am J Dermatopathol 1994; 16 (5): 542–548.
Sterry W, Hauschild A . Loss of leucocyte common antigen (CD45) on atypical lymphocytes in the localized but not disseminated type of Pagetoid reticulosis. Br J Dermatol 1991; 125 (3): 238–242.
Smoller BR, Bishop K, Glusac EJ, Kim YH, Bhargava V, Warnke RA . Reassessment of lymphocyte immunophenotyping in the diagnosis of patch and plaque stage lesions of mycosis fungoides. Appl Immunohistochem 1995; 3 (1): 32–36.
Smoller BR, Bishop K, Glusac EJ, Bhargava V, Kim YH, Warnke RA . Lymphocyte antigen abnormalities in inflammatory dermatoses. Appl Immunohistochem 1995; 3: 127–131.
Yagi H, Hagiwara T, Shirahama S, Tokura Y, Takigawa M . Disseminated pagetoid reticulosis: need for long-term follow-up. J Am Acad Dermatol 1994; 30: 345–349.
Berti E, Aversa GG, Soligo D, Cattoretti G, Delia D, Aiello A, et al. A6—a new 45RO monoclonal antibody for immunostaining of paraffin-embedded tissues. Am J Clin Pathol 1991; 95 (2): 188–193.
Alaibac M, Chu AC . Pagetoid reticulosis: a γ/δ T-cell lymphoma? Eur J Dermatol 1992; 2: 109–111.
Ioannides G, Engel MF, Rywlin AM . Woringer-Kolopp disease (pagetoid reticulosis). Am J Dermatopathol 1983; 5 (2): 153–158.
Tan RS, MacLeod TI, Dean SG . Pagetoid reticulosis, epidermotropic mycosis fungoides and mycosis fungoides: a disease spectrum. Br J Dermatol 1987; 116 (1): 67–77.
Zackheim HS . Is “localized epidermotropic reticulosis” (Woringer-Kolopp disease) benign? J Am Acad Dermatol 1984; 11: 276–283.
Resnik KS, Kantor GR, Lessin SR, Kadin ME, Chooback L, Cooper HS, et al. Mycosis fungoides palmaris et plantaris. Arch Dermatol 1995; 131 (9): 1052–1056.
Oliver GF, Winkelmann RK . Unilesional mycosis fungoides: a distinct entity. J Am Acad Dermatol 1989; 20 (1): 63–70.
McNiff JM, Schechner JS, Crotty PL, Glusac EJ . Mycosis fungoides palmaris et plantaris or acral pagetoid reticulosis? Am J Dermatopathol 1998; 20 (3): 271–275.
Smoller BR . Mycosis fungoides palmaris et plantaris. Arch Dermatol 1996; 132: 468.
Mandojana RM, Helwig EB . Localized epidermotropic reticulosis (Woringer-Kolopp disease). J Am Acad Dermatol 1983; 8 (6): 813–829.
Shapiro PE, Pinto FJ . The histologic spectrum of mycosis fungoides/Sezary syndrome (cutaneous T-cell lymphoma). A review of 222 biopsies, including newly described patterns and the earliest pathologic changes. Am J Surg Pathol 1994; 18 (7): 645–667.
Dummer R, Michie SA, Kell D, Gould JW, Haeffner AC, Smoller BR, et al. Expression of bcl-2 protein and Ki-67 nuclear proliferation antigen in benign and malignant cutaneous T-cell infiltrates. J Cutan Pathol 1995; 22 (1): 11–17.
Kaudewitz P, Burg G, Majiewski G, Gerdes J, Schwarting R, Braun FO . Cell populations in pagetoid reticulosis: an immunological study using cell activation associated monoclonal antibodies. Acta Derm Venereol 1987; 67 (1): 24–29.
Luther H, Bacharach BM, Schultz EU, Altmeyer P . Pagetoid reticulosis of the Ketron-Goodman type. Hautarzt 1989; 40 (8): 530–535.
Cerroni L, Rieger E, Hodl S, Kerl H . Clinicopathologic and immunologic features associated with transformation of mycosis fungoides to large-cell lymphoma. Am J Surg Pathol 1992; 16 (6): 543–552.
Stein H, Mason DY, Gerdes J, O’Connor N, Wainscoat J, Pallesen G, et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985; 66 (4): 848–858.
Acknowledgements
The authors thank Drs. Elizabeth Abel, Todd Anhalt, Jeff Herten, Jane Kardashian, Lia Keller, James McCuster, and Eric Wall for clinical information.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Haghighi, B., Smoller, B., LeBoit, P. et al. Pagetoid Reticulosis (Woringer-Kolopp Disease): An Immunophenotypic, Molecular, and Clinicopathologic Study. Mod Pathol 13, 502–510 (2000). https://doi.org/10.1038/modpathol.3880088
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880088
