
Abstract
Two cases of synovial sarcoma that arose in the upper digestive tract are reported. One case was a polypoid mass that arose at the gastroesophageal junction; the other was a large intramural mass that arose in the wall of the stomach. Both cases had a classic biphasic pattern. In the stomach tumor, the biphasic morphology was focal and there was an abrupt transition to poorly differentiated synovial sarcoma. The tumors had immunohistochemical features that were consistent with synovial sarcoma. Ultrastructural evaluation of the gastroesophageal tumor supported the diagnosis. The diagnostic X;18 translocation was demonstrated by fluorescence in situ hybridization on sections from paraffin-embedded tissue in 86% and 50% of interphase nuclei from the gastroesophageal and gastric tumor, respectively. The translocation was present in equal frequency in the epithelial and spindle cells in the biphasic areas and the poorly differentiated areas of the gastric tumor, indicating that the development of the more aggressive subclone was probably due to genetic mutations not encompassing the SYT-SSX gene fusion product. We are aware of only five reported cases of synovial sarcoma arising in the digestive tract, all in the proximal esophagus. These cases are the first reported arising in the gastroesophageal junction and stomach and the only cases of synovial sarcoma of the digestive tract in which the diagnostic translocation was demonstrated. Sarcomatoid carcinoma (carcinosarcoma) and gastrointestinal stromal tumor are the main differential diagnoses for synovial sarcoma in this site. Synovial sarcoma of the digestive tract may be underdiagnosed, and its recognition may have important clinical implications. Fluorescence in situ hybridization is helpful in making this distinction.
Similar content being viewed by others

INTRODUCTION
Synovial sarcomas are malignant mesenchymal tumors of uncertain histogenesis. They may be biphasic, monophasic, or poorly differentiated. They usually arise in the extremities intimately related to tendons and bursal structures of large joints but rarely in the synovium. Occasionally, they arise in the head and neck, most commonly as retropharyngeal or parapharyngeal masses (1).
We describe two cases of synovial sarcoma that arose in the upper digestive tract. One case originated at the gastroesophageal junction, and the other arose in the stomach. We are aware of five previously reported cases of primary synovial sarcoma of the cervical esophagus (2, 3, 4, 5, 6), but, to our knowledge, synovial sarcoma involving gastroesophageal junction or stomach has not been previously reported. Both tumors displayed histologic features of synovial sarcoma. Immunohistochemical staining profiles supported the diagnosis. Ultrastructural features of the gastroesophageal junction tumor were consistent with synovial sarcoma. However, because of the unusual locations, cytogenetic studies were performed to confirm the diagnoses. The characteristic X;18 translocation was demonstrated by using fluorescence in situ hybridization (FISH) studies on paraffin-embedded tissue.
MATERIALS AND METHODS
Formalin-fixed, paraffin-embedded tissue was sectioned at 4 μm and stained with hematoxylin and eosin, and periodic-acid-Schiff with and without diastase predigestion according to standard methods.
For immunohistochemical examination, sections from both tumors were stained with monoclonal antibodies directed against epithelial membrane antigen (EMA; Ventana, Tucson, AZ), vimentin (Ventana), CD56 (Novocastra, Newcastle upon Tyne, UK), CD57 (Becton-Dickinson, San Jose, CA), and CD99 (O13; Signet, Dedham, MA). Monoclonal antibodies directed against cytokeratin AE1/3 (DAKO, Carpinteria, CA) and wide-spectrum antikeratin polyclonal antibodies (DAKO) were used on the gastroesophageal tumor. A cocktail of cytokeratin AE1/3 (DAKO) and CAM5.2 (DAKO) was used on the gastric tumor. Antibody dilutions were individually optimized (Table 1) for use with the Ventana automatic slide stainer (Ventana). Microwave pretreatment was used for antibodies directed against EMA, vimentin, CD56, CD57, CD99, and the AE1/3 and CAM 5.2 cocktail. Appropriate tissue served as positive controls. Negative controls were carried out with the Ventana Medical Systems negative control reagent, mouse monoclonal antibody Clone MOPC-21′, which is not directed against a known epitope present in human tissue.
For ultrastructural evaluation, tissue was fixed in 3% glutaraldehyde, postfixed in osmium tetroxide, and stained with uranyl acetate and lead citrate. The specimen was thin sectioned and examined under a transmission electron microscope.
Cytogenetic evaluation was performed using FISH with dual color DNA probes, a green probe for the X centromere and a red probe for a region in the middle of the 18 long-arm (18q21). Hybridization with probes DXZ1 (Oncor, Gaithersburg, MD) labeled with fluoroisothiocyanate for the X centromere and digoxigenin-labeled 18qD18541 detected with rhodamine (Oncor) were performed as previously described (7). Briefly, this involved deparaffinization, digestion with pronase for 30 min, denaturation of specimen DNA in 70% formamide at 70° C, separate denaturation of probe DNA in a microwave oven for 40 seconds, and hybridization of probe and tumor DNA overnight in a 37° C humidified chamber according to the manufacturer's instructions. Slides from the gastroesophageal synovial sarcoma and an adenocarcinoma of the lung were processed together, and 100 cells from each were scored blindly. Slides from the gastric tumor were processed in the same fashion with an adenocarcinoma of the stomach as a control. If the X;18 translocation is present, one would expect a high frequency of spatial association between the X and 18 signals that would otherwise be randomly distributed within the nucleus. Because we are unaware of any report of an X;18 translocation in lung or gastric cancer, this tissue was used to control for the frequency of X and 18 signals that might be closely associated by chance. A close association was defined as the distance between a red and green signal less than the width of three centromere signals. Random associations were defined as signals more than or equal to three centromere widths. Using this methodology, we have previously found that the frequency of close associations in synovial sarcoma generally exceeded 75% compared with 20% or less of tumor nuclei lacking an X;18 translocation (7).
CASE REPORTS
Case 1
A 47-year-old Hispanic man had a history of persistent peptic ulcer disease and pyloric stenosis associated with gastric outlet obstruction. He had undergone repeated esophagogastroduodenoscopies with pyloric dilatations and standard medical therapy without significant improvement. As a result of the refractory nature of his peptic ulcer disease and gastric outlet obstruction, he underwent vagotomy, antrectomy, and a Billroth II procedure.
During surgery, a pedunculated, polypoid mass was found arising at the gastroesophageal junction. The mass was excised and submitted as a separate specimen. Histologic examination of the polypoid mass revealed a biphasic tumor consistent with synovial sarcoma. The tumor was focally present at the surgical margin. The patient underwent a subsequent completion gastrectomy with partial esophagectomy. Residual synovial sarcoma was not identified.
No adjuvant therapy was given. Follow-up evaluation including roentgenograms, computerized tomographic (CT) scans, and frequent esophagojejunostomy endoscopic procedures revealed no evidence of other primary site, metastasis, or recurrence. The patient has remained disease-free for 21 months.
Case 2
A 55-year-old woman had a 3-year history of abdominal swelling. Approximately 4 weeks before presentation, she developed dull right-side abdominal pain, nausea, vomiting, and rectal bleeding. The patient's medical history was noncontributory. An endoscopic biopsy revealed a poorly differentiated neoplasm interpreted as consistent with a malignant gastrointestinal stromal tumor (GIST). An abdominal CT scan demonstrated a large mass in the right upper quadrant between the gastric antrum, left lobe of the liver, duodenum, and pancreas. A chest CT scan demonstrated emphysematous change and a small peripheral nodule in the upper lobe of the right lung, which was too small to biopsy. No serum elevations of carcinoembryonic antigen, CA 19–9, CA 125, or alpha-fetoprotein were noted.
During laparotomy, a large mass arising from the distal stomach and adherent to the left lobe of the liver, gallbladder, and retroperitoneum overlying the pancreas was found. Multiple liver metastases were present. A hemigastrectomy with retrocolic Billroth II procedure was performed to debulk the tumor. No evidence of another primary site has been found. The patient died of disease 6 months after her resection.
RESULTS
Macroscopic Findings
The tumor at the gastroesophageal junction was a 5.2 × 2.2 × 1.3-cm, pedunculated, tan-brown, soft, smooth-surface mass. The gastric mass was a 16 × 15 × 7.5-cm spherical, intramural mass covered by a focally ulcerated gastric epithelium. On cut section, it had a fleshy, tan-pink appearance and large areas of necrosis.
Light Microscopic Findings
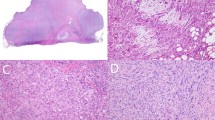
The gastroesophageal tumor showed the classic biphasic pattern of synovial sarcoma composed of epithelial cells in a sarcomatous spindle cell background (Fig. 1). The epithelial cells were arranged in discrete, simple to complex, gland-like structures and focally solid nests. The gastric tumor focally exhibited biphasic morphology (Fig. 2). The gland-like structures of both tumors contained eosinophilic, periodic-acid-Schiff–positive, diastase-resistant, amorphous material. The epithelial cells in both lesions had uniform, round to oval, vesicular nuclei with occasional, small nucleoli and relatively abundant eosinophilic cytoplasm. The spindle cell component of the biphasic region in both tumors varied from densely cellular to paucicellular to myxoid. The spindle cells had plump, fusiform nuclei with fine, granular chromatin, inconspicuous nucleoli, and lightly basophilic to eosinophilic cytoplasm. Scattered mast cells and neutrophils were present in the background. The majority of the gastric tumor was poorly differentiated, appearing as a high-grade sarcoma, with an abrupt transition from the biphasic to poorly differentiated areas in the tumor (Fig. 2). The poorly differentiated areas were predominantly composed of sheets of round to oval, hyperchromatic tumor cells admixed with some loose, myxoid areas. In the gastroesophageal tumor, the mitotic rate was low with an average of one mitotic figure per 10 high power fields. In the gastric tumor, the mitotic rate was variable, with an average of 9 mitotic figures per 10 high power fields in the biphasic portion of the tumor but greater than 50 mitotic figures per 10 high power fields in the poorly differentiated areas, including many atypical mitotic figures.

The gastroesophageal tumor was composed of well-demarcated, gland-like structures in a spindle cell background.

A, low power view of the gastric tumor demonstrating extension of the tumor into the superficial subepithelial stroma. B, there was an abrupt transition from the focal, biphasic area (top) to the predominant, poorly differentiated areas of the gastric tumor (bottom).
Immunohistochemistry
The immunohistochemical results are summarized in Table 1. The epithelial cells were positive for cytokeratin (AE1/3, wide-spectrum polyclonal cytokeratin, or AE1/3 and CAM 5.2 cytokeratin cocktail), whereas the spindle cells showed only rare, focal staining. The glands exhibited luminal positivity for EMA. The spindle cells in both tumors were positive for vimentin and the neural crest adhesion molecule CD56, whereas the epithelial cells were negative; in the stomach lesion, the staining for vimentin was somewhat patchy. The gastric tumor was focally positive for CD57, whereas the gastroesophageal tumor was negative. Both tumors were negative for CD99 (O13). The poorly differentiated areas of the gastric tumor had a similar immunohistochemical staining profile as the spindle cell component of the biphasic area. The poorly differentiated component exhibited the same staining pattern for the mesenchymal and neural antigens but failed to exhibit the focal staining for cytokeratin seen in the spindle cell component of the biphasic portion of the tumor.
Ultrastructural Examination
Ultrastructural examination was performed on the gastroesophageal tumor. Electron micrographs showed a distinct, biphasic cell population with epithelial-like cells forming glandular, tubular, or solid structures and elongated stromal cells (Fig. 3). The epithelial-stromal junction was composed of a discrete electron-dense basal lamina. The nuclei in the epithelial cells tended to be round to oval, and the nuclei in the stromal cells tended to be more spindle shaped. Both were irregularly shaped with coarse, clumped, marginated chromatin. The cytoplasm of the epithelial cells had tubular mitochondria, Golgi apparatus, lysosomal bodies, and lipid vacuoles. Junctional complexes were present between adjoining cells. The luminal surface had microvilli and focal cytoplasmic filaments. No basal lamina or cellular attachment was noted between stromal cells, which appeared surrounded by fibrillar ground substance as well as mature collagen.

Ultrastructural examination of the gastroesophageal tumor demonstrated a distinct biphasic cell population separated by a discrete electron dense basal lamina. The epithelial cells have numerous microvilli projecting into a luminal space (magnification, 4950 ×).
Cytogenetic Results with FISH
Approximately two thirds of the cells could not be scored because of overlapping, truncation, or unclear signals. In the scorable cells, fluorescent signals for the X centromere and the 18q21 region were found to be closely associated in 86% and 50% of the interphase nuclei in the gastroesophageal and gastric tumor, respectively, indicating the presence of the X;18 translocation (Fig. 4). In the gastroesophageal tumor, the epithelial and spindle cells demonstrated the translocation with equal frequency. Similarly, the translocation was present with equal frequency in both components of the biphasic portion of the gastric tumor as well as in the poorly differentiated area. The control slide from the adenocarcinoma of the lung demonstrated a close association in 22% of tumor nuclei. This is somewhat higher than expected. However, the lung tumor had four X chromosomes, thereby increasing the chance of random close associations of chromosomes X and 18. The control slide from the adenocarcinoma of the stomach demonstrated close associations of the X and 18 chromosomes in 2% of the cells.

Typical appearance of the red X and green 18 probes characterizing the diagnostic t(X;18) in four separate synovial sarcoma nuclei from the gastroesophageal tumor (A) and the gastric tumor (B). The translocation is characterized by the red and green signals touching or lying less than three centromere lengths to each other (lower left corner of A) in the interphase nuclei. The second green 18 signal is randomly located in the nuclei. The second red X signal is randomly located in the gastric tumor, but there is no second red X signal in the gastroesophageal tumor because the patient is male. This pattern is consistent with the presence of the X;18 translocation.
DISCUSSION
Primary sarcomas of the esophagus and stomach are rare. Fewer than 2% of primary esophageal tumors are mesenchymal in origin (8); the majority of esophageal sarcomas are leiomyosarcoma (9). In the stomach, the majority of malignant mesenchymal tumors include malignant gastric stromal tumors and leiomyosarcoma. To our knowledge, there have been only five previous case reports of synovial sarcomas arising in the digestive tract, all of which involved the cervical or proximal esophagus (Table 2). Similar to our gastroesophageal junction case, these lesions all were exophytic masses exhibiting classic biphasic histology. We are unaware of any previously reported case of synovial sarcoma arising in the stomach. Our cases are the first involving the digestive tract in which the characteristic X;18 translocation was identified.
The primary differential diagnosis of biphasic synovial sarcoma in the digestive tract is sarcomatoid carcinoma (carcinosarcoma). The age ranges for both tumors overlap; however, sarcomatoid carcinoma tends to present at an older age (10, 11) than synovial sarcoma (1). Sarcomatoid carcinomas of the esophagus (11, 12, 13, 14, 15, 16) and stomach (12, 17, 18, 19) usually present as polypoid masses with a similar gross appearance as our tumor from the gastroesophageal junction. Gastric sarcomatoid carcinomas also frequently present as intramural masses (12), grossly indistinguishable from our case arising in the stomach. Carcinosarcomas of the esophagus are most common in the middle third of the esophagus (11), whereas gastric sarcomatoid carcinomas have a predilection for the pyloric stomach (12), although they have been reported in a variety of sites within the respective organs. Previously reported cases of esophageal synovial sarcomas have been restricted to the proximal esophagus (2, 3, 4, 5, 6).
The sarcomatous elements in esophageal or gastric sarcomatoid carcinoma may have a variety of histologic features. Typically, they have been undifferentiated spindle cells similar in appearance to synovial sarcoma, but overt mesenchymal differentiation showing smooth muscle, cartilage, or bone formation has been described (12, 17, 19, 20). Mesenchymal differentiation can occur in synovial sarcoma as well.
Most esophageal sarcomatoid carcinomas have a squamous epithelial component (6, 11, 12, 14, 20, 21, 22), although in rare cases adenocarcinoma or adenoid cystic carcinoma occurs (20). The lack of squamous carcinoma in our case makes the diagnosis of esophageal sarcomatoid carcinoma less likely. In gastric sarcomatoid carcinoma, adenocarcinoma is the predominant epithelial component (9, 12, 17, 18, 19). The epithelial component can vary from well differentiated to poorly differentiated, including signet ring morphology (9, 12), but well differentiated adenocarcinoma is more typical (10). The neoplastic glands can blend with the stromal component or be well demarcated from it; similar patterns are seen in biphasic synovial sarcoma. Differentiating between synovial sarcoma and gastric sarcomatoid carcinoma is therefore more problematic. In our opinion, the epithelial cells of our cases were somewhat more bland with more well-demarcated and complex glandular structures than in typical gastric sarcomatoid carcinoma. However, the histologic features of sarcomatoid carcinoma, particularly gastric tumors, can be strikingly similar.
The differential diagnosis of monophasic fibrous or poorly differentiated synovial sarcomas of the digestive tract includes benign and malignant GIST and leiomyosarcoma. In fact, the stomach lesion was originally interpreted as consistent with a malignant GIST on the basis of a biopsy that showed only the poorly differentiated areas of the tumor and none of the biphasic component. The presence of the biphasic area caused us to question the original diagnosis.
Though synovial sarcoma can have a distinctive appearance, establishing a definitive diagnosis may require ancillary studies, especially in tumors that arise in unusual locations. By immunohistochemistry, the epithelial and mesenchymal elements of sarcomatoid carcinoma and synovial sarcoma can have essentially the same pattern of reactivity with antibodies directed against epithelial markers and vimentin. However, synovial sarcoma often expresses neuroectodermal antigens (23, 24, 25). CD99 expression has been demonstrated in 46 to 100% of synovial sarcomas (23). Folpe et al. (24) described immunoreactivity for CD56, the neural crest adhesion molecule, and CD57, a marker of nerve sheath differentiation, in 8 of 8 and 9 of 10 poorly differentiated synovial sarcomas, respectively. Immunostains for CD56 were positive in the poorly differentiated part of the gastric tumor and selectively marked the spindle cell component in the biphasic components of both tumors, as previously reported in biphasic synovial sarcoma (25). The gastric tumor showed focal positivity for CD57, whereas the gastroesophageal junction tumor was negative. Both were negative for CD99. Theoretically, a sarcomatoid carcinoma with neural differentiation could display a similar immunohistochemical profile, although we are not aware of a case of gastric or esophageal sarcomatoid carcinoma that expressed these neural antigens. Given the similar immunostaining profile of the spindle cell and poorly differentiated component of the gastric tumor, we interpreted the poorly differentiated area as evolving from the less prominent biphasic component. In more equivocal cases in which GIST would still be a consideration in the differential diagnosis, immunohistochemistry may be of use as GISTs are usually positive for CD34, unlike synovial sarcoma (26). The stomach tumor was negative for smooth muscle actin and desmin (data not shown).
The ultrastructural features support the diagnosis of synovial sarcoma but are not specific. Ultrastructural examination of esophageal (14) and gastric sarcomatoid carcinoma (9, 17, 18) has shown similar features as found in synovial sarcoma (1).
Because of the unusual locations of the tumors, we decided to pursue cytogenetic confirmation. FISH conclusively established the presence of the X;18 translocation, a sensitive marker demonstrated in 70 to 90% of synovial sarcomas (27, 28, 29). The specific t(X;18)(p11.2;q11.2) results in the fusion gene product SYT-SSX (30). Only rare nonsynovial sarcomas have exhibited an X;18 translocation, including a single case of fibrosarcoma (31), a case of malignant fibrohistiocytoma (32), a case of poorly differentiated sarcoma not otherwise specified (32), and four cases of malignant peripheral nerve sheath tumor (33). This translocation has not, to our knowledge, been described in carcinosarcomas of the esophagus or stomach. Loss of heterozygosity at different loci has been described in esophageal carcinosarcoma, but there was no evidence of an X;18 translocation (13). Because of its relative specificity, the X;18 translocation may prove useful in the diagnosis of synovial sarcoma in unusual locations. In the gastric synovial sarcoma, the percentage of cells with the translocation was slightly less than expected (50%) but still significantly more than would be expected by chance. The poorly differentiated portion of the gastric tumor demonstrated the translocation in the same proportion as the biphasic areas. Therefore, the development of the more aggressive subclone is likely due to additional genetic mutations not encompassing the gene fusion product of the translocation. It would be interesting to investigate whether this finding holds true for other cases of synovial sarcoma that exhibit poorly differentiated areas admixed with more typical biphasic and monophasic morphology.
The clinicopathologic characteristics of our cases and the five previously reported cases of synovial sarcoma arising in the esophagus are summarized in Table 2. All of the tumors involving the esophagus had an exophytic appearance variably described as papillary or polypoid. Our case arising in the stomach was intramural and spherical. Reported cases of digestive tract synovial sarcoma have a broad age range (14 to 75 years); the majority of the tumors occurred in younger individuals. Of the five patients with esophageal tumors, four were alive without disease after resection, with follow-up ranging from 20 to 48 months; one patient died of disease 2 years after diagnosis. The patient with the gastric synovial sarcoma died of disease 6 months after resection. The follow-up from previous reports and our case indicates that esophageal or gastroesophageal synovial sarcomas may have a better prognosis than synovial sarcoma that arises elsewhere. Symptoms from luminal obstruction could result in earlier detection, or other unknown factors may play a role. Theoretically, the opposite may be true for tumors that arise in the stomach because they could grow to large size before obstructive or other mass-related symptoms occur. Clinical follow-up has been limited, and synovial sarcoma is widely known to recur or metastasize after long disease-free intervals. Illustrative of this point is the 5- and 10-year overall survival of 60% and 34%, respectively, for synovial sarcoma (34). Singer et al. (34) found that synovial sarcomas smaller than 5 cm had 100% 10-year survival. It is interesting that the only reported death as a result of esophageal synovial sarcoma occurred with a tumor 2.5 cm in greatest dimension (6). This suggests that synovial sarcoma may behave differently in different sites. The 5-year survival rate of esophageal sarcomatoid carcinoma ranges from approximately 27 to 50% (11, 12), which is significantly worse than synovial sarcoma. Gastric sarcomatoid carcinoma has a much worse prognosis, approaching a mortality of 100% at 5 years (12). Distinguishing synovial sarcoma from sarcomatoid carcinoma at the gastroesophageal junction or stomach may therefore have important clinical implications in terms of patient prognosis. A retrospective cytogenetic examination of previously diagnosed sarcomatoid carcinomas to identify unrecognized synovial sarcomas might help clarify the prognostic implications. The distinction has therapeutic implications as well. Synovial sarcoma may respond to adjuvant chemotherapy with regimens including high-dose ifosfamide (35, 36).
Synovial sarcoma of the digestive tract is possibly an underdiagnosed condition. Only a handful of cases have been reported, yet we have seen two cases in the past 2 years. Some cases may have been previously diagnosed as sarcomatoid carcinoma or GIST. As cytogenetic and molecular techniques become more accessible, the accurate diagnosis of synovial sarcoma that arises in the digestive tract may increase.
References
Enzinger FM, Weiss SW . Synovial sarcoma. In: Soft tissue tumors. 3rd ed. St. Louis: Mosby; 1995. pp. 757–786.
Amr SS, Shihabi NK, Al Hajj H . Synovial sarcoma of the esophagus. Am J Otolaryngol 1984; 5: 266–269.
Antón-Pacheco J, Cano I, Cuadros J, Vilariño A, Berchi F . Synovial sarcoma of the esophagus. J Pediatr Surg 1996; 31: 1703–1705.
Bloch MJ, Iozzo RV, Edmunds LH Jr, Brooks JJ . Polypoid synovial sarcoma of the esophagus. Gastroenterology 1987; 92: 229–233.
Habu S, Okamoto E, Toyosaka A, Nakai Y, Takeuchi M . Synovial sarcoma of the esophagus: report of a case. Surg Today 1998; 28: 401–404.
Palmer BV, Levene A, Shaw HJ . Synovial sarcoma of the pharynx and oesophagus. J Laryngol Otol 1983; 97: 1173–1176.
Zilmer M, Harris CP, Steiner DS, Meisner LF . Use of nonbreakpoint DNA probes to detect the t(X;18) in interphase cells from synovial sarcoma: implications for detection of diagnostic tumor translocations. Am J Pathol 1998; 152: 1171–1177.
Sons HU, Borchard F . Esophageal cancer: autopsy findings in 171 cases. Arch Pathol Lab Med 1984; 108: 983–988.
Robey-Cafferty SS, Grignon DJ, Ro JY, Cleary KR, Ayala AG, Ordonez NG, et al. Sarcomatoid carcinoma of the stomach: a report of three cases with immunohistochemical and ultrastructural observations. Cancer 1990; 65: 1601–1606.
Cho KJ, Myong NH, Choi DW, Jang JJ . Carcinosarcoma of the stomach: a case report with light microscopic, immunohistochemical, and electron microscopic study. APMIS 1990; 98: 991–995.
Iyomasa S, Kato H, Tachimori Y, Watanabe H, Yamaguchi H, Itabashi M . Carcinosarcoma of the esophagus: a twenty-case study. Jpn J Clin Oncol 1990; 20: 99–106.
Iezzoni JC, Mills SE . Sarcomatoid carcinomas (carcinosarcomas) of the gastrointestinal tract: a review. Sem Diagn Pathol 1993; 10: 176–187.
Iwaya T, Maesawa C, Tamura G, Sato N, Ikeda K, Saski A, et al. Esophageal carcinosarcoma: a genetic analysis. Gastroenterology 1997; 113: 973–977.
Ooi A, Kawahara E, Okada Y, Mizukami Y, Sugawara S, Noto Y, et al. Carcinosarcoma of the esophagus: an immunohistochemical and electron microscopic study. Acta Pathol Jpn 1986; 36: 151–159.
Osamura RY, Shiamura K, Hata J, Tamaoki N, Watanabe K, Kubota M, et al. Polypoid carcinoma of the esophagus: a unifying term for “carcinosarcoma” and “pseudosarcoma.” Am J Surg Pathol 1978; 2: 201–208.
Perch SJ, Soffen EM, Whittington R, Brooks JJ . Esophageal sarcomas. J Surg Oncol 1991; 48: 194–198.
Dundas SA, Slater DN, Wagner BE, Mills PA . Gastric adenocarcinoleiomyosarcoma: a light, electron microscopic and immunohistological study. Histopathology 1988; 13: 347–350.
Hanada M, Nakano K, Ii Y, Takami M . Carcinosarcoma of the stomach: a case report with light microscopic, immunohistochemical, and electron microscopic study. Acta Pathol Jpn 1985; 35: 951–959.
Siegal A, Freund U, Gal R . Carcinosarcoma of the stomach. Histopathology 1988; 13: 350–353.
Stener B, Kock NG, Pettersson S, Zetterlund B . Carcinosarcoma of the esophagus. J Thorac Cardiovasc Surg 1967; 54: 746–750.
Taniyama K, Sasaki N, Mukai T, Uemura N, Miyoshi N, Nakai H, et al. Carcinosarcomas of the esophagus. Pathol Int 1995; 45: 297–302.
Xu L, Sun C, Wu L, Chang Z, Liu T . Clinical and pathological characteristics of carcinosarcoma of the esophagus: report of four cases. Annu Thorac Surg 1984; 37: 197–203.
Dei Tos AP, Wadden C, Calonje E, Sciot R, Pauwels P, Knight JC, et al. Immunohistochemical demonstration of glycoprotein p30/32MIC2 (CD99) in synovial sarcoma: a potential cause for diagnostic confusion. Appl Immunohistochem 1995; 3: 168–173.
Folpe AL, Schmidt RA, Chapman D, Gown AM . Poorly differentiated synovial sarcoma: immunohistochemical distinction from primitive neuroectodermal tumors and high-grade malignant peripheral nerve sheath tumors. Am J Surg Pathol 1998; 22: 673–682.
Miettinen M, Cupo W . Neural cell adhesion molecule distribution in soft tissue tumors. Hum Pathol 1993; 24: 62–66.
Suster S, Fisher C, Moran CA . Expression of bcl-2 oncoprotein in benign and malignant spindle cell tumors of soft tissue, skin, serosal surfaces, and gastrointestinal tract. Am J Surg Pathol 1998; 22: 863–872.
Limon J, Mrozek K, Mandahl N, Nedoszytko B, Verhest A, Rys J, et al. Cytogenetics of synovial sarcoma: presentation of ten new cases and review of the literature. Genes Chrom Cancer 1994; 7: 502–508.
Sreekantaiah C, Ladanyi M, Rodriguez E, Chaganti RSK . Chromosomal aberrations in soft tissue tumors: relevance to diagnosis, classification, and molecular mechanisms. Am J Pathol 1994; 144: 1121–1134.
van de Rijn M, Barr FG, Collins MH, Xiong QB, Fisher C . Absence of SYT-SSX fusion products in soft tissue tumors other than synovial sarcoma. Am J Clin Pathol 1999; 112: 43–49.
Clark J, Rocques PJ, Crew AJ, Gill S, Shipley J, Chan AML, et al. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet 1994; 7: 502–508.
Limon J, Dal Cin P, Sandberg AA . Translocations involving the X chromosome in solid tumors: presentation of two sarcomas with t(X;18)(q13;p11). Cancer Genet Cytogenet 1986; 23: 87–91.
Mandahl N, Heim S, Arheden K, Rydholm A, Willén H, Mitleman F . Multiple karyotyping rearrangements, including t(X; 18)(p11;q11), in a fibrosarcoma. Cancer Genet Cytogenet 1988; 30: 323–327.
Balazs L, Parham D, Shurtleff S, Pappo A, Downing J . Identification of the t(X;18) as a diagnostic tool in the differential diagnosis of synovial sarcoma versus peripheral nerve sheath tumors [abstract]. Mod Pathol 1997; 10: 8.
Singer S, Baldini EH, Demetri GD, Fletcher JA, Corson JM . Synovial sarcoma: prognostic significance of tumor size, margin of resection, and mitotic activity for survival. J Clin Oncol 1996; 14: 1201–1208.
Kampe CE, Rosen G, Eilber F, Eckardt J, Lowenbraun S, Foster J, et al. Synovial sarcoma: a study of intensive chemotherapy in 14 patients with localized disease. Cancer 1993; 72: 2161–2169.
Rosen G, Forscher C, Lowenbraun S, Eilber F, Eckardt J, Holmes C, et al. Synovial sarcoma: uniform response of metastases to high dose ifosfamide. Cancer 1994; 73: 2506–2511.
Acknowledgements
The authors thank Drs. James W. Smith and John N. Eble for their generous support, Dr. Christopher D.M. Fletcher for reviewing Case 1, Dr. John D. Henley for reviewing the manuscript, Peggy Harger-Allen for her assistance with the electron microscopy, and Jeanne Bordeaux-Heller for performance of FISH analyses.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Billings, S., Meisner, L., Cummings, O. et al. Synovial Sarcoma of the Upper Digestive Tract: A Report of Two Cases with Demonstration of the X;18 Translocation by Fluorescence In Situ Hybridization. Mod Pathol 13, 68–76 (2000). https://doi.org/10.1038/modpathol.3880011
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880011
Keywords
This article is cited by
-
Laparoscopic intragastric resection of gastric synovial sarcoma: report of the first ever case with video demonstration
World Journal of Surgical Oncology (2021)
-
Primary monophasic synovial sarcoma of the cervical esophagus confirmed by detection of the SS18-SSX2 fusion transcripts: case report and literature review
Surgical Case Reports (2020)
-
Diffuse, Aggressive Metastatic Progression after Minimally Invasive Local Resection of Primary Gastric Synovial Sarcoma: a Case Report and Systematic Review of the Literature
Journal of Gastrointestinal Cancer (2019)
-
Clear Cell Sarcoma-Like Tumor of the Gastrointestinal Tract
Journal of Gastrointestinal Cancer (2019)
-
Establishment and proteomic characterization of a novel synovial sarcoma cell line, NCC-SS2-C1
In Vitro Cellular & Developmental Biology - Animal (2018)
