
Abstract
We report the development of a reverse transcriptase polymerase chain reaction assay that reliably detects the ETV6-NTRK3 chimeric RNA characteristic of infantile fibrosarcoma and the cellular variant of congenital mesoblastic nephroma (CMN) in formalin-fixed, paraffin-embedded tissue blocks. The 188 base pair polymerase chain reaction fusion product was detected in 11 of 12 cases of cellular CMN from which a larger sized control RNA band could be amplified, and even in 7 of 8 cases in which the control band was not detectable. A variety of other tumors that are in the histologic differential diagnosis of cellular CMN yielded negative results, including four classic CMNs, four rhabdoid tumors of the kidney, and four clear cell sarcomas of the kidney, confirming the assay’s specificity. We further demonstrate the assay’s utility by illustrating two cases of molecularly confirmed cellular CMN that mimicked rhabdoid tumor and clear cell sarcoma of the kidney. In contrast to previous reports, five mixed CMNs that had both classic and cellular areas all lacked the ETV6-NTRK3 fusion transcript. These results suggest that cases morphologically defined as mixed CMN may represent a mixed group of genetically distinct entities.
Similar content being viewed by others
INTRODUCTION
The discoveries of specific chromosomal translocations associated with many pediatric soft-tissue tumors represent major advances toward understanding the molecular pathogenesis of these lesions (1). The chimeric RNAs produced from the gene fusions can be detected by reverse transcriptase polymerase chain reaction (RT-PCR), creating sensitive and specific diagnostic assays for each of these tumors. Typically, such analysis requires fresh tissue that yields intact RNA. The adaptation of these RT-PCR assays to RNA extracted from formalin-fixed, paraffin-embedded tissue (2, 3, 4) has made these tests applicable to virtually all specimens and has helped to more sharply define the clinical and pathologic spectra of many of these tumors.
A novel t(12;15)(p13;q25) translocation that results in an ETV6-NTRK3 gene fusion was recently discovered in infantile fibrosarcoma (5). Of note, this fusion was not found in a limited number of cases of either adult type fibrosarcoma or infantile fibromatosis. Subsequently, two groups (6, 7) have demonstrated the identical ETV6-NTRK3 gene fusion in the cellular variant of congenital mesoblastic nephroma (CMN) of the kidney, which is morphologically identical to infantile fibrosarcoma and affects the same age group. The classical variant of CMN, which is morphologically identical to infantile fibromatosis, did not contain the fusion transcript. These results strongly support that cellular CMN represents infantile fibrosarcoma of the kidney and renal sinus and indicate that the classical variant of CMN represents a distinct entity. It is interesting that a small number of cases of “mixed” CMN, which have both cellular and classical foci, were reported to be fusion positive (6, 7).
Because of the tremendous morphologic overlap that exists among pediatric renal tumors and the diagnostic dilemmas thereby created, and to analyze a larger number of mixed CMN cases, we attempted to adapt an RT-PCR assay for the ETV6-NTRK3 gene fusion to formalin-fixed, paraffin-embedded tissue. We report the development of this assay and its application to problematic renal tumor cases and to mixed CMN cases.
MATERIALS AND METHODS
Case Selection
The files of the National Wilms Tumor Study Pathology Center and the personal consultation files (OCWT files) of one of the authors (JBB) were searched for cases of CMN or cases in which CMN was a strong diagnostic consideration but for which another diagnosis had been rendered. From this group of more than 300 cases, those for which a paraffin-embedded, formalin-fixed tissue block was available (approximately 30) were selected. All slides were reviewed by one author (PA). Cases for which an alternative diagnosis seemed possible and all cases of mixed cellular and classical mesoblastic nephroma were reviewed together by two of the authors (PA and JBB) using a dual observer microscope. Clinical data were gleaned from the submitting institution’s pathology report.
RNA Extraction
A detailed description of this method has been previously published (3); the following summary includes minor modifications from that method. Briefly, four 10-μ sections from each tissue block were cut and placed in a sterile Eppendorf tube. Microtome blades were changed between cutting each block. After two xylene washes for deparaffinization and two ethanol washes to remove xylene, the tissue was resuspended in 0.35 mL of digestion buffer (20 mm Tris pH 8.0, 10 mm EDTA, 1% sodium dodecyl sulfate [SDS] in diethylpyrocarbonate-treated, autoclaved water) and digested with 500 μg proteinase K overnight at 55° C. The next day, RNA was extracted using 0.9 mL of phenol/guanidine isothiocyanate (Trizol; Gibco BRL, Friendswood, TX) and 0.2 mL chloroform followed by vortexing. After centrifugation, the upper aqueous phase was removed; placed in a new Eppendorf tube; and precipitated with 0.7 mL isopropanol, 75 μl of 3 m sodium acetate, and 2 μl of Pellet Paint Co-Precipitant (Novagen, Madison, WI). After successive 70% and 100% ethanol washes, the pellet was resuspended in 30 μL of diethylpyrocarbonate-treated water, treated with DNase I (Gibco BRL), and frozen at −20° C until use.
Reverse Transcriptase Polymerase Chain Reaction
Approximately one tenth (4 μL) of each RNA extract was reverse transcribed in a volume of 20 μl with 50 pmol random hexamers, 100 units of Superscript II Reverse Transcriptase (Gibco BRL), 2 mm each nucleotide, 5 mm magnesium chloride, and 20 units RNase inhibitor (Boehringer Mannheim, Indianapolis, IN) in PCR buffer (Perkin-Elmer, Norwalk, CT). The reaction was performed at 42° C for 30 min in a Coy Tempcycler II thermocycler, Model 110 S (Coy Corp., Grass Lake, MI). The enzyme and first-strand cDNA were then denatured at 94° C for 5 min and chilled on ice for 5 min. To assess the adequacy of the RNA, the entire 20 μL RT reaction was subjected to PCR using primers (2) (Table 1) that span an intron of the ubiquitously expressed phosphoglycerate kinase gene (PGK). Because the primers chosen span an intron, the product of amplification of any contaminating DNA would be approximately 200 base pairs larger than and hence readily distinguishable from the product of RNA amplification (263 base pairs). Because PGK has no known processed pseudogenes, the ability to produce this 263 base pair product indicates that intact, amplifiable RNA fragments of at least that size are still present in the block, despite processing. A negative (water) control was run with each set of cases. The PCR cycling parameters were as follows: 1 min each at 95, 66, 72, 95, 65, 72, 95, 64, 72, 95, 63, 72, 95, 62, 72, 95, 61, and 72° C, followed by 35 cycles at 95, 60, and 72° C for 1 min each. A 10-min final extension at 72° C ended the protocol. Twenty-five microliters of each PCR reaction (approximately one fourth) underwent electrophoresis in 2.5% agarose gels containing 50% Nu-Sieve Agarose (FMC Bioproducts, Rockland, ME) and 50% Ultrapure agarose (Gibco BRL), and was visualized with ethidium bromide staining.
To detect the ETV6-NTRK3 fusion transcript, we designed primers (Table 1) that closely flanked the published fusion breakpoint, so as to amplify a relatively small 188 base pair PCR product. This strategy has previously proved effective in detecting chimeric RNA transcripts among the partially degraded RNA present in tissue blocks (2, 3, 4). The primers chosen were designed and optimized using the Oligo Primer Analysis Software Version 5.0 (Molecular Biology Insights, Inc., Cascade, CO), and BLAST analysis (http://www.ncbi.nih.gov) was performed to rule out significant primer annealing with other known genes. Reverse transcription, PCR, and electrophoresis conditions were identical to those used with the PGK primers. A negative (water) control and a known positive control block were run with every set of cases tested. All primers were synthesized at the Johns Hopkins University School of Medicine DNA Analysis Facility.
Southern Blotting and Oligonucleotide Probing
After the products of the ETV6-NTRK3 PCR reactions were visualized by ethidium bromide staining, these agarose gels were soaked briefly in distilled water, then transferred onto Hybond-N+ charged nylon membranes (Amersham, Buckinghamshire, England) using 0.4 mm sodium hydroxide as the transfer buffer. After transfer, membrane pH was neutralized using two 5-min washes of 2 × standard sodium citrate (SSC), and membranes were stored at room temperature.
Membranes were hybridized to a labeled oligonucleotide probe that spans the last 13 ETV6 bases and the first 11 NTRK3 bases at the fusion point of the chimeric RNA transcript (Table 1). A chemiluminescence detection system was used as follows. One hundred picomoles of the fusion probe was 3′-end labeled with Digoxigenin using the DIG Oligonucleotide 3′-end labeling kit (Boehringer Mannheim, Indianapolis, IN) according to the manufacturer’s instructions and stored at −20° C until used. Each membrane was placed in Dig Easy-Hyb solution (Boehringer) for 1 hour at 42° C as a prehybridization step to block nonspecific binding. The probe was boiled for 5 min, placed on ice for 2 min, added to 10 mL of DIG Easy-Hyb solution, and hybridized to the membrane overnight at 42° C.
The next day, membranes were washed twice at low (2 × SSC, 0.1% SDS) and high (0.1% SSC, 0.1% SDS) stringencies for 10 min at 42° C. Each membrane was washed in 1 × DIG Wash buffer (Boehringer) for 1 to 5 min at room temperature, then blocked with 1 × DIG Blocking buffer for 30 min at room temperature. Following the manufacturers instructions for the DIG Luminescent Detection Kit for Nucleic Acids (Boehringer), an antidigoxigenin Fab antibody fragment conjugated to alkaline phosphatase was diluted 1:10,000 into 1 × Blocking buffer and placed on the membrane for 30 min at room temperature. Each membrane was washed twice with 1 × Wash buffer for 15 min at room temperature, then equilibrated with 1 × Detection buffer. CSPD substrate (disodium-3-[4-methoxyspiro{1,2-dioxetane-3, 2′-(5′-chloro) ticyclo(3.3.1.1) decan}-4-y1]phenyl phosphate) was diluted 1:100 into 1 × Detection buffer and placed on the membrane for 5 min at room temperature. Excess CSPD was then drained from the membrane, which was sealed in plastic wrap and placed at 37° C for 10 min. Chemiluminescent signal was detected by exposure of the membrane to Hyperfilm ECL high-performance chemiluminescent film (Amersham) for periods of 10 seconds (short exposure), 2 min, and 20 min (long exposure).
RESULTS
Assay Verification
RNA extracted from a known case of CMN was subjected to RT-PCR using the designed ETV6-NTRK3 primer pair to test its performance. The reaction produced a sharp band easily visible on an ethidium bromide stained agarose gel and consistent with 188 base pair predicted size. To verify its identity, the remainder of this PCR product was purified with the HighPure PCR product purification kit (Boehringer) and directly sequenced in forward and reverse directions using the ETV6 forward and NTRK3 reverse primers, respectively, on an ABI PRISM Model 377 automated sequencer (PE Biosystems, Foster City, CA). A sequence precisely matching the ETV6/NTRK3 fusion reported in the literature (5) was obtained (GenBank accession number of junction sequence: AF041811)(data not shown).
Assay Performance
The ETV6-NTRK3 gene fusion was detected in 11 of 12 available blocks of CMN from which a PGK band could be produced and hence for which RNA integrity was verifiable (Fig. 1). These 11 blocks represented tumors from 10 different children: the additional positive block was an abdominal recurrence of one child’s tumor. Cytogenetic reports were available on two of these cases: both showed trisomy 11, but in neither case was the t(12;15)(p13;q25) detected. Cases with adequate RNA (PGK positive) that tested negative for the fusion transcript included four classic CMN, four clear cell sarcomas of the kidney, four rhabdoid tumors of the kidney, three synovial sarcomas (one of which was a primary renal tumor), one metanephric stromal tumor, one cystic nephroma with infiltrative edges that mimicked CMN, one ossifying renal tumor of infancy with CMN-like areas, one cystic hamartoma of the renal pelvis (for which the submitted diagnosis was “adult mesoblastic nephroma”), one anaplastic sarcoma not otherwise specified, one alveolar rhabdomyosarcoma, one intra-abdominal desmoplastic small round cell tumor, and one pleuropulmonary blastoma.

Detection of ETV6-NTRK3 fusion in congenital mesoblastic nephroma. Top, agarose gel electrophoresis of PGK RT-PCR products reveals the presence of a 263 base pair band signifying amplifiable RNA in all cases except for Case 7. The size of selected bands in the molecular weight marker lane M are indicated on the left. Middle, agarose gel electrophoresis of reverse transcriptase polymerase chain reaction products obtained with ETV6-NTRK3 primers on the same RNAs. Cases 1 to 4 are cellular congenital mesoblastic nephromas (CMNs), with Case 3 representing the cellular CMN with prominent rhabdoid features illustrated in Figure 2. Case 5 is a mixed CMN for which the block studied showed only classic morphology, whereas both Cases 6 and 7 are classic CMNs. Bottom, Southern blot of the agarose gel in the middle panel hybridized with the ETV6-NTRK3 fusion probe confirms the positivity of Cases 1 to 4 and the negativity of Cases 5 to 7. Note that Case 7 is not truly evaluable because the phosphoglycerate kinase gene result was negative.
Of note, among eight blocks of cellular CMNs from which a PGK band could not be obtained and, hence, from which the RNA quality was questionable, an ETV6-NTRK3 fusion product could still be detected in seven. One of these blocks was from a case in which the first block tested was both PGK positive and fusion positive. Another block contained less than 10% tumor in a background of normal kidney tissue but still proved to be fusion positive. To exclude cDNA contamination as a cause for the fusion transcript positivity in the cases with a negative PGK result, a control PCR reaction in which reverse transcriptase was omitted was run with each of these RNAs when they were tested for the ETV6-NTRK3 fusion and was appropriately negative. The ETV6-NTRK3 fusion was not detected in either of the two pure classic mesoblastic nephromas from which a PGK band could not be detected.
In all cases, the identity of the PCR product obtained was confirmed by Southern blotting and oligonucleotide fusion probe hybridization. All appropriately sized intense bands hybridized to the probe. Cases judged to be negative by ethidium bromide staining proved negative on hybridization with the fusion probe, even after prolonged 20-min (10-fold excess) exposure.
Application to Problematic Cases
Case 1: Cellular CMN versus Rhabdoid Tumor of Kidney
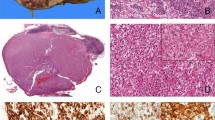
A 14-month-old boy presented with a unilateral renal tumor and hypercalcemia. This high-grade lesion invaded the renal vein but appeared completely excised. On light microscopy, part of the tumor showed a plump, spindle cell appearance with moderately sized nucleoli compatible with cellular CMN. However, much of the tumor consisted of cells with rhabdoid features consisting of vesicular nuclei, macronucleoli, and eccentric cytoplasm suggesting an inclusion (Fig. 2). This part of the tumor aggressively invaded the renal parenchyma as a rhabdoid tumor typically does. Immunohistochemistry (not reviewed) was reported by the submitting pathologist to show staining of the same cells with vimentin, desmin, and low molecular weight cytokeratin. Of note, sections taken from a portion of the tumor that was implanted into a nude mouse showed purely rhabdoid features. The submitted diagnosis favored rhabdoid tumor of the kidney. On review, the favored diagnosis was cellular CMN with rhabdoid features.

Cellular mesoblastic nephroma mimicking rhabdoid tumor of kidney. A, intermediate power (200×) view of one portion of tumor showing fascicles of spindled cells with pale cytoplasm and inconspicuous nucleoli, typical of cellular congenital mesoblastic nephroma. B, intermediate power view (200×) of the tumor’s invasive edge, showing diffuse infiltration of the renal parenchyma by more hyperchromatic, epithelioid-appearing cells with prominent nucleoli. Note the entrapped benign renal tubule at the upper left. C, high power (630×) view of this area, showing remarkable cytologic resemblance of this area to rhabdoid tumor of the kidney. Reverse transcriptase polymerase chain reaction confirmed the presence of the ETV6-NTRK3 gene fusion in this case (see lane 3, Fig. 1). Hematoxylin and eosin stained sections.
As shown in Figure 1, the ETV6-NTRK3 fusion transcript was detected in the available tissue block by RT-PCR, confirming the diagnosis of CMN. On clinical follow-up, the patient developed a solitary lung metastasis 1 year later that was resected. He subsequently remains free of disease 7 years later.
Case 2: Cellular CMN versus Clear Cell Sarcoma of the Kidney
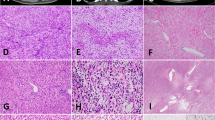
An 11-month-old male underwent nephrectomy for a 8.5-cm, 980-g, partially necrotic renal tumor. The tumor was largely consistent with CMN, as it contained intersecting fascicles of plump spindle cells that pushed aside the kidney. However, less cellular areas featured sclerosis, entrapment of tubules and glomeruli, and a prominent capillary vascular pattern, all features suggestive of clear cell sarcoma of the kidney (CCSK) (Fig. 3). RT-PCR detected the ETV6-NTRK3 fusion transcript in the available block, confirming the diagnosis of CMN.

Cellular congenital mesoblastic nephroma (CMN) mimicking clear cell sarcoma of the kidney. A, intermediate (200×) power view of one portion of the tumor, showing dense spindle cell fascicular pattern typical of CMN. B, more hypocellular region of the same tumor at intermediate power (200×) showing sclerosis, myxoid change, capillary vasculature, and entrapment of native renal tubules that suggests clear cell sarcoma of the kidney. This tumor also tested positive for the ETV6-NTRK3 fusion by reverse transcriptase polymerase chain reaction, confirming the diagnosis of cellular CMN. Hematoxylin and eosin stained sections.
Mixed Classic and Cellular Mesoblastic Nephroma
Paraffin blocks were obtained from nine cases of mixed cellular and classic mesoblastic nephroma. Of these, two failed to yield amplifiable RNA (PGK negative), leaving seven cases for study. For five of these seven cases, an available block containing both a cellular and a classic component was studied. The proportion of cellular tumor ranged from 5 to 80% in these blocks. In two of these five cases, an additional available block showing only the classical area was also studied. None of these blocks yielded the ETV6-NTRK3 fusion transcript, an absence confirmed by oligonucleotide hybridization (Fig. 4). In addition, microdissection of slides cut from four of the five mixed blocks was performed to isolate the classic from the cellular components for RNA extractions. None of these areas (all of which yielded amplifiable RNA) bore the fusion transcript (data not shown).

Analysis of mixed mesoblastic nephromas for ETV6-NTRK3 fusion transcript. Top, agarose gel electrophoresis of PGK reverse transcriptase polymerase chain reaction products show that all 5 mixed congenital mesoblastic nephromas (CMNs) and the positive control(+), a block from a cellular CMN, yielded adequate RNA. Lanes 1 and 2 are different blocks from the same tumor. The sizes of selected bands in the molecular weight marker lane are indicated on the right. Middle, agarose gel electrophoresis of ETV6-NTRK3 reverse transcriptase polymerase chain reaction products of the same cases. Whereas the positive control yielded a distinct 188 base pair band, none of the mixed CMN cases yielded a similar result. Bottom, Southern blot of the middle panel gel probed with the ETV6-NTRK3 fusion probe, confirming the negativity of the mixed CMN cases in the presence of a positive cellular CMN case control.
Paraffin blocks were available from only the classic area of the two remaining mixed mesoblastic nephromas. Both of these blocks were negative for the ETV6/NTRK3 fusion.
DISCUSSION
We report the development and application of an RT-PCR assay that detects the ETV6-NTRK3 fusion transcript characteristic of both infantile fibrosarcoma and cellular CMN in formalin-fixed, paraffin-embedded tissue blocks. The assay proved to be extremely sensitive, detecting the fusion in 11 of 12 blocks of cellular CMN that yielded amplifiable RNA. Specificity was verified by negative results with a variety of other pediatric renal tumors and sarcomas and confirmed by oligonucleotide probe hybridization. An added measure of the tests’ sensitivity was that a fusion product could be detected in seven of eight blocks from which the control RNA band (PGK) could not be amplified. This likely relates in part to the smaller amplicon size of the ETV6-NTRK3 fusion product (188 base pairs) in this assay as compared with the control RNA amplicon (263 base pairs). Because the RNA extracted from formalin-fixed, paraffin-embedded tissue blocks is typically degraded, there is likely a size cut-off over which an RNA segment cannot be amplified. The cut-off for these fusion-positive, PGK-negative cases was likely somewhere between 263 and 188 base pairs. Another factor that determines the success of amplification is the level of expression and stability of each specific transcript. Given that the ETV6-NTRK3 gene fusion, like other sarcoma-associated gene fusions, is thought to be the primary genetic alteration in this neoplasm, it is likely that the fusion transcript is expressed at high levels, making its detection easier. Indeed, the fusion transcript has been detectable on routine Northern blotting analysis (5). One potential limitation of our PCR assay is that it may not detect possible variations in ETV6-NTRK3 fusion transcript structure, because the primers chosen are very close to the usual fusion point. Such variation has been well documented in other sarcoma-associated gene fusions, such as the EWS/FLI 1 gene fusion of Ewing sarcoma (8), and limits the sensitivity of these RT-PCR assays. Such variation has not been reported in the ETV6-NTRK3 chimera.
The difficulty of the differential diagnosis that cellular CMN can pose and the therapeutic consequences of these diagnoses demonstrate the clinical utility of this assay. Both rhabdoid tumor of the kidney and cellular CMN usually affect young children younger than 2 years, are centered in the renal medulla, and have been associated with hypercalcemia (9). Although rhabdoid tumor cells are usually ovoid, a spindled fascicular pattern that mimics cellular CMN has been described and illustrated (10). As the case we have illustrated shows, cellular CMN, which typically has moderately sized nucleoli, can have strikingly enlarged nucleoli that overlap those of rhabdoid tumor of the kidney. The potential for misdiagnosis in this case was amplified by the reported immunohistochemical results that suggested a polyphenotypic pattern of staining typical of rhabdoid tumor. The distinction of cellular CMN from rhabdoid tumor of the kidney is of utmost importance to these young patients. Cellular CMN is cured with 95% certainty by complete resection; patients do not usually receive adjuvant therapy and are followed by monthly ultrasonography for 1 year before being pronounced cured (11). In contrast, rhabdoid tumor has a dismal prognosis—80% of children die within 18 months (10). As such, a diagnosis of rhabdoid tumor of the kidney prompts intensive chemotherapy and radiation therapy that is particularly toxic to the infant population commonly afflicted. Treatment-related deaths are not uncommon. Hence, a misdiagnosis of a cellular CMN as a rhabdoid tumor of the kidney could result in unnecessary patient toxicity or even death. Confirmation of the histologic and molecular diagnoses of cellular CMN was provided by this patient’s clinical course, which was more consistent with that of a cellular CMN than that of rhabdoid tumor. CMN, typically locally aggressive, can metastasize to distant organs such as lung and brain (12, 13). Long-term survival after resection of recurrences is documented (J.B. Beckwith, unpublished observations). In contrast, long-term survival from metastatic rhabdoid tumor would be nearly unfathomable.
Whereas CCSK is more common after the first year of life in which CMN predominates, clinical overlap with CMN exists particularly in the infant of near 1 year of age. Grossly, both tumors are again typically centered in the renal medulla, and both are commonly cystic. Whereas cellular CMN is again usually spindled, the plump cell variant features more ovoid cells with prominent capillary vasculature, which can simulate that of CCSK. CCSK can also show a spindled pattern that mimics cellular CMN. The distinction can usually be made by the coarser chromatin and presence of nucleoli in cellular CMN but can be obscured by inadequate fixation. This distinction is again clinically relevant, because CCSK should be treated with chemotherapeutic regimens containing doxorubicin. The specificity of the presence of the fusion transcript for cellular CMN that we demonstrate is concordant with the results of Knezevich et al. (6), who found it to be absent in 12 cases of CCSK.
The absence of the ETV6-NTRK3 fusion transcript in the mixed CMN cases we studied was an unexpected result, given the two previous studies (6, 7) that found the fusion transcript in three mixed cases and ETV6 rearrangements by fluorescence in situ hybridization in two others (7). It is difficult to reconcile our results with the results of these previous studies. It is conceivable that our mixed cases contained the t(12;15)(p13;q25) translocation but did not express the fusion transcript at high enough levels to be detectable in our paraffin-based assay. This possibility seems unlikely given the high sensitivity of our assay for the presence of fusion transcript in pure cellular CMN, as exemplified by our ability to detect the fusion in the absence of detectable PGK. However, one could always postulate that mixed CMN express the fusion transcript at lower levels and that this might underlie their morphologic heterogeneity. Regardless of the explanation, our assay does not seem to be useful for the molecular diagnosis of mixed CMN.
Taking all of the molecular and morphologic evidence together, however, we believe that the morphologic appearance of mixed CMN likely represents more than one genetic entity. First, some of these cases, including some of those shown in this study to test negative for the ETV6-NTRK3 fusion transcript, seem to be classic CMN that have small cellular foci indistinguishable from those of true cellular CMN. These are cases in which the cellular areas are multifocal and patchy and blend imperceptibly with the classic areas that form the majority of the tumor (Fig. 5). We would view these cases as analogues to the well-documented occurrence in soft tissue of infantile fibromatosis with cellular areas that overlap morphologically with infantile fibrosarcoma. We would not expect the morphologic transitions seen in such cases to be accompanied by a clonal genetic event such as a translocation, because they are not sharply demarcated. Second, other “mixed” CMNs that are fusion positive might actually be pure cellular tumors with foci of artificially decreased cellularity that simulate the classic pattern. In support of this hypothesis, we found one of the tumors initially coded as a mixed CMN to be fusion positive; on review, we reclassified this neoplasm as a cellular CMN with focal less cellular zones at its periphery likely resulting from compression and edema. This case highlights that the histologic distinction of cellular and classic CMN is not always clear cut. Cellular tumors usually have pushing borders, prominent mitoses, necrosis, and increased nuclear density, whereas classic CMNs are more infiltrative, are less mitotically active, lack necrosis, and produce more collagen. However, borderline cases—tumors with prominent mitoses but infiltrative borders, tumors of intermediate cellularity—exist and are difficult to classify morphologically. These cases can confound the diagnosis of mixed CMN. Finally, it is likely that some true mixed CMNs contain the t(12;15)(p13;q25) translocation. It is tempting to speculate, as Rubin et al. (7) have, that these tumors have the translocation throughout but acquire trisomies (particularly at chromosome 11) specifically within their cellular areas, because the acquisition of trisomies in the cellular foci of mixed CMN has previously been documented (14, 15, 16). However, the presence of the fusion transcript in a classical area of a mixed CMN has not been demonstrated in either this or either of the previous studies. Another possibility is that these mixed tumors acquire the translocation during progression from classic morphology to cellular morphology, though sarcoma-associated translocations have never been documented to be progression events in neoplasia. Further studies using other modalities, such as fluorescence in situ hybridization or immunohistochemistry, will be required to determine definitively the presence and distribution of the ETV6/NTRK3 fusion transcript in mixed CMN.

Intermediate power view of a mixed congenital mesoblastic nephroma that tested negative for the ETV6-NTRK3 fusion transcript in this study. The cellular foci are of the “blue cell” type in this tumor and merge imperceptibly with the classic pattern. Hematoxylin and eosin stained section, 100×.
Accession codes
References
Ladanyi M . The emerging molecular genetics of sarcoma translocations. Diagn Mol Pathol 1995; 4: 162–173.
Argani P, Perez-Ordonez B, Xiao H, Caruana SM, Huvos AG, Ladanyi M . Olfactory neuroblastoma is not related to the Ewing family of tumors: absence of EWS/FLI1 gene fusion and MIC2 expression. Am J Surg Pathol 1998; 22: 391–398.
Argani P, Zakowski MF, Klimstra DS, Rosai J, Ladanyi M . Detection of the SYT-SSX chimeric RNA of synovial sarcoma in paraffin-embedded tissue and its application in problematic cases. Mod Pathol 1998; 11: 65–71.
Edwards RH, Chatten J, Xiong QB, Barr FG . Detection of gene fusions in rhabdomyosarcoma by reverse transcriptase-polymerase chain reaction assay of archival samples. Diagn Mol Pathol 1997; 6: 91–97.
Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH . A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet 1998; 18: 184–187.
Knezevich SR, Garnett MJ, Pysher TJ, Beckwith JB, Grundy PE, Sorensen PH . ETV6-NTRK3 gene fusions and trisomy 11 establish a histogenetic link between mesoblastic nephroma and congenital fibrosarcoma. Cancer Res 1998; 58: 5046–5048.
Rubin BP, Chen CJ, Morgan TW, Xiao S, Grier HE, Kozakewich HP, et al. Congenital mesoblastic nephroma t(12;15) is associated with ETV6-NTRK3 gene fusion: cytogenetic and molecular relationship to congenital (infantile) fibrosarcoma. Am J Pathol 1998; 153: 1451–1458.
Zucman J, Melot T, Desmaze C, Ghysdael J, Plougastel B, Peter M, et al. Combinatorial generation of variable fusion proteins in the Ewing family of tumours. EMBO J 1993; 12: 4481–4487.
Murphy WM, Beckwith JB, Farrow GM . Tumors of the kidney, bladder, and related urinary structures. Atlas of Tumor Pathology. Third Series, Fascicle 11. Washington DC: Armed Forces Institute of Pathology; 1994. pp. 55–67, 82–89.
Weeks DA, Beckwith JB, Mierau GW, Luckey DW . Rhabdoid tumor of kidney: a report of 111 cases from the National Wilms’ Tumor Study Pathology Center. Am J Surg Pathol 1989; 13: 439–458.
Beckwith JB . Renal tumors. In: Stocker JT, Askin FB, editors. Pathology of solid tumors in children. New York: Chapman & Hall Medical; 1998. pp. 16–20.
Vujanic GM, Delemarre JF, Moeslichan S, Lam J, Harms D, Sandstedt B, et al. Mesoblastic nephroma metastatic to the lungs and heart—another face of this peculiar lesion: case report and review of the literature. Pediatr Pathol 1993; 13: 143–153.
Heidelberger KP, Ritchey ML, Dauser RC, McKeever PE, Beckwith JB . Congenital mesoblastic nephroma metastatic to the brain. Cancer 1993; 72: 2499–2502.
Kovacs G, Szucs S, Maschek H . Two chromosomally different cell populations in a partly cellular congenital mesoblastic nephroma. Arch Pathol Lab Med 1987; 111: 383–385.
Schofield DE, Yunis EJ, Fletcher JA . Chromosome aberrations in mesoblastic nephroma. Am J Pathol 1993; 143: 714–724.
Pettinato G, Manivel JC, Wick MR, Dehner LP . Classical and cellular (atypical) congenital mesoblastic nephroma: a clinicopathologic, ultrastructural, immunohistochemical, and flow cytometric study. Hum Pathol 1989; 20: 682–690.
Acknowledgements
We are indebted to Helen Fedor for preparing sections for RNA extraction and to Lisa Madden for typing the manuscript. We thank the following pathologists for providing tissue blocks for this study: Roger Williams, M.D. (Oakland, CA), Laura Finn, M.D. (Seattle, WA), and Kathleen Heidelberger, M.D. (Ann Arbor, MI). Special thanks are due to Nancy Browning, whose organization of the National Wilms Tumor Study Pathology Center allowed this study and others to be performed. The National Wilms Tumor Study Group is supported primarily by United States Public Health Service Grant no. CA-42386.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Argani, P., Fritsch, M., Kadkol, S. et al. Detection of the ETV6-NTRK3 Chimeric RNA of Infantile Fibrosarcoma/Cellular Congenital Mesoblastic Nephroma in Paraffin-Embedded Tissue: Application to Challenging Pediatric Renal Stromal Tumors. Mod Pathol 13, 29–36 (2000). https://doi.org/10.1038/modpathol.3880006
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3880006
Keywords
This article is cited by
-
Japanese Society of Medical Oncology/Japan Society of Clinical Oncology/Japanese Society of Pediatric Hematology/Oncology-led clinical recommendations on the diagnosis and use of tropomyosin receptor kinase inhibitors in adult and pediatric patients with neurotrophic receptor tyrosine kinase fusion-positive advanced solid tumors
International Journal of Clinical Oncology (2023)
-
Multidisciplinary consensus on optimising the detection of NTRK gene alterations in tumours
Clinical and Translational Oncology (2021)
-
Clinical implications of intratumor heterogeneity: challenges and opportunities
Journal of Molecular Medicine (2020)
-
Methods for Identifying Patients with Tropomyosin Receptor Kinase (TRK) Fusion Cancer
Pathology & Oncology Research (2020)
-
Expression of cyclin D1 in clear cell sarcoma of kidney. Is it useful in differentiating it from its histological mimics?
Diagnostic Pathology (2019)
