
Abstract
Large-cell neuroendocrine and small-cell lung carcinomas are highly aggressive neuroendocrine tumors that can be associated in a variant of ‘small-cell lung carcinoma combined with large-cell neuroendocrine carcinoma’. Little is known about this rare tumor type with biphenotypic neuroendocrine differentiation. The aim of the present study was to genetically characterize each component of a series of combined small-cell/large-cell neuroendocrine carcinomas, to gain information on their histogenesis and to compare the alterations observed with those found in their respective pure forms. To this end, 22 formalin-fixed, paraffin-embedded lung neuroendocrine tumors obtained from surgical resections were investigated: six combined small-cell/large-cell carcinomas, eight pure large-cell carcinomas and eight pure small-cell carcinomas. For the combined neuroendocrine neoplasms, DNA was extracted separately from each of the two cytologically different populations. Allelic imbalance was investigated by PCR amplification of 30 highly polymorphic microsatellite markers located at 11 different chromosomal regions. A common background of genetic alterations, similar in both components of the combined neoplasms, was demonstrated at 17p13.1, 3p14.2–3p21.2, 4q12–4q24, 5q21 and 9p21. In fact, the two components appeared to be more similar to each other than to their respective pure forms. In addition, allelic imbalances preferentially involving one of the two components were found. These alterations often appeared to be specific for this histological variant, as compared with those observed in pure forms or in the literature. In conclusion, this is the first report in which a molecular characterization of the variant of small-cell lung carcinoma combined with large-cell neuroendocrine carcinoma was performed. The finding of common alterations in the two phenotypically different neuroendocrine cell components suggests a close genetic relationship and supports the hypothesis of a monoclonal origin from a common ancestor. The genetic differences observed provide the basis for the divergent differentiation and parallel the morphological differences in the two components of these combined neuroendocrine neoplasms.
Similar content being viewed by others


Main
Neuroendocrine tumors of the lung are a family of neoplasms arising from the embryological foregut, which share common neuroendocrine markers plus the presence of neuroendocrine granules at electron microscopy, but which differ greatly in their biological behavior. They include typical and atypical carcinoids, with respectively low and intermediate malignant potential, and large-cell neuroendocrine and small-cell lung carcinomas, with a highly malignant clinical course and a dismal prognosis.1, 2
The distinction between large- and small-cell carcinomas is based substantially on cytological features, including larger vs smaller cell size, abundant vs scant cytoplasm, the presence vs absence of prominent nucleoli, coarsely vs finely granular chromatin, and polygonal vs round to fusiform shapes, respectively.3 However, a histological differential diagnosis between these two aggressive forms of neuroendocrine tumors can be difficult in some cases, due mostly to a frequent overlapping of cell size.4, 5 On a molecular level, both similarities and differences have been described.6, 7, 8 While some authors have emphasized the distinctive features separating large- and small-cell carcinomas,6, 7 others have underlined similarities, and called for their inclusion within the single group of high-grade neuroendocrine tumors.4, 8
The only variant of small-cell lung carcinoma described in the latest WHO classifications of lung neoplasms is the ‘combined small-cell lung carcinoma’, in which small-cell carcinoma is associated with an additional non-small-cell component.9 This category also includes small-cell carcinomas combined with large-cell neuroendocrine carcinomas, but this is a rare variant that has not been extensively studied, particularly as regards the genetic alterations occurring in each of the two neuroendocrine components.
The aim of the present study was to separately analyze the genetic alterations characterizing each tumor component in a series of combined small-cell/large-cell neuroendocrine carcinomas of the lung. Both chromosomal regions harboring genes commonly involved in the pathogenesis of lung carcinomas, and regions reported to be more selectively involved in large-cell neuroendocrine or small-cell lung carcinoma were investigated. For comparison, the same analysis was also performed in a series of pure high-grade neuroendocrine lung tumors.
Materials and methods
Tumors
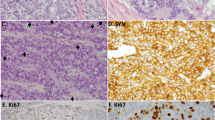
A total of 22 formalin-fixed and paraffin-embedded pulmonary neuroendocrine tumors obtained from surgical resections were investigated: six combined small-cell/large-cell neuroendocrine carcinomas, eight pure large-cell neuroendocrine carcinomas and eight pure small-cell lung carcinomas (Table 1). Informed consent was obtained from all patients and/or guardians, in accordance with Italian law. All tumors were examined independently by three pathologists who are expert in the field (G Pelosi, C Bordi, and G Rindi), classified according to the histopathological criteria proposed by the WHO classification9 (Figure 1), and staged according to the latest TNM system (TNM, 6th edition, UICC, 2002). In the event of disagreement, a final consensus diagnosis was reached.

(a) Pure large-cell neuroendocrine carcinoma (LCNEC) of the lung, with cells characterized by abundant cytoplasm and prominent nucleoli (H&E, × 200). (b) Pure small-cell lung carcinoma (SCLC), with cells characterized by searee cytoplasm and small, round to oval, nuclei with inconspicuous nucleoli (H&E, × 200). (c) Combined small-cell/large-cell neuroendocrine carcinoma characterized by clear-cut separation of the two tumor-cell components (H&E, × 150).
The combined neuroendocrine neoplasms were characterized by a clear-cut separation of the two cell populations (Figure 1c), thus allowing a molecular analysis of each neoplastic component to be performed. Both tumor and non-tumor tissues (ie, adjacent histologically normal lung parenchyma or uninvolved regional lymph nodes) were available.
DNA Extraction
Normal and tumor areas were manually microdissected from serial 4-μm-thick histological sections stained with hematoxylin. The two cell populations of combined neuroendocrine neoplasms were microdissected separately in order to extract DNA from each single tumoral component. A neoplastic cellularity of at least 80% was obtained for all tumor samples. DNA extraction and purification were performed using a commercial kit (DNeasy Tissue kit; QIAgen Inc., Valencia, CA, USA).
PCR Amplification and Fragment Analysis
Allelic imbalance was investigated with 30 highly polymorphic microsatellite markers (Table 2) located at 11 chromosomal regions (3p, 4q, 5q, 6p, 6q, 9p, 10q, 16q, 17p, X–Y pseudoautosomal regions 1 and 2). Microsatellite markers were chosen according to two different criteria: some located in chromosomal regions commonly altered in lung carcinomas (3p, 5q, 9p, 16q, 17p), while others in regions reported to be potentially discriminating between small-cell and large-cell neuroendocrine carcinoma (4q, 5q33, 6p, 10q, X).6, 7, 10, 11 All tumors from female patients had previously been investigated for X chromosome loss of heterozygosity.11 To verify the involvement of X chromosome in male patients, microsatellites located in the homologous regions at both ends of sex chromosomes (X–Y pseudoautosomal regions, X–Y PAR 1 and 2) were investigated. Microsatellite markers were PCR-amplified from tumor and control DNA using primers labelled with Beckman Coulter WellRED fluorescent dyes D3 or D4 (Beckman Coulter, Fullerton, CA, USA). For PCR amplification, 2 μl of DNA were combined in a 25-μl reaction mixture containing 10 mM Tris-HCl (pH 9), 50 mM KCl, 0.1% Triton X-100, 200 μM of each dNTP (Promega, Madison, WI, USA), 0.4 μM of each primer, 1.5–2.0 mM MgCl2 and 1.25 U Taq polymerase (Promega). PCR amplifications were performed with an AB 2700 (Applied Biosystems, Foster City, CA, USA) thermal cycler for 35 cycles. Individual PCR products were run on an eight-capillary CEQ™8000 DNA sequencer (Beckman Coulter), as previously described.11 CEQ8000 Fragment Analysis software (Beckman Coulter) generated electrophoretic profiles in which alleles appeared as peaks, with area and height proportional to the concentration of PCR fragments.
Evaluation of Allelic Imbalance
An asymmetric allelic ratio in the tumor DNA at a microsatellite locus, as compared with the ratio observed in normal DNA, was described as allelic imbalance. Heterozygosity, that is, the presence of two distinct alleles in the normal tissue, was the prerequisite for evaluation of allelic imbalance. Peak height values produced by the Fragment Analysis software were used to calculate the following ratio: (lower allele/higher allele)TUM/(lower allele/higher allele)NORM. Allelic imbalance, scored when the ratio values were <0.6 or >1.67, may reflect the complete loss of one allele masked by the presence of a background of normal cells, by tumor heterogeneity or by non-clonal loss, or it may reflect an increased DNA copy number.12 For each tumor case, a fractional allelic imbalance index was calculated as follows: number of chromosomal regions harboring at least one allelic imbalance/number of chromosomal regions with at least one informative marker.
Genetic Concordance in Combined Neuroendocrine Neoplasms
Genetic concordance was defined as the finding of an absence of alterations or the presence of imbalances involving the same allele in both components of each combined neuroendocrine tumor.
Fluorescent In Situ Hybridization
Six male patients' tumors displaying allelic imbalance at X–Y PAR regions (combined tumor nos. 4 and 5, large-cell neuroendocrine carcinoma nos. 13 and 14, small-cell lung carcinoma nos. 21 and 22) were investigated by fluorescent in situ hybridization (FISH) as described elsewhere,13 in order to understand which sex chromosome was affected by the imbalance. The chromosome enumeration probe (CEP) used was a mixture of a spectrum orange-labelled CEP X probe specific for the α-satellite centromeric region of chromosome X, and a spectrum green-labelled CEP Y probe specific for the satellite III (Yq12) region of chromosome Y (Vysis Inc., Doweners Grove, IL, USA). The absence of orange or green spots in the tumors was indicative of loss of chromosome X or Y, respectively.
Statistical Analysis
Frequencies of allelic imbalance were compared using Fisher's exact test. Fractional allelic imbalance indexes were compared with the non-parametric Mann–Whitney test. All analyses were carried out using GraphPad InStat version 3.06 for Windows (GraphPad Software, San Diego, CA, USA). All P-values were based on two-sided testing. P-values less than 0.05 were considered statistically significant.
Results
Allelic Imbalances in Combined Neuroendocrine Carcinomas (n=6)
Allelic imbalances involving both components of combined small-cell/large-cell neuroendocrine carcinomas with the same frequency were found at 17p (p53) in 100% of cases (6/6); at 3p (RASSF1A, FHIT) and 4q in 83% of cases (5/6) and at 5q (APC) and 9p (CDKN2A and CDKN2B) in 50% of cases (3/6) (Figure 2a; Table 2). Allelic imbalances preferentially involving one of the two components were also found, although these differences did not reach statistical significance. The large-cell component was more frequently altered than the small-cell component at 10q (83 vs 50%), 16q (83 vs 67%) and 6q (80 vs 40%). The small-cell component was more frequently altered at X–Y PAR2 (67 vs 50%), X–Y PAR1 (50 vs 33%) and 6p (40 vs 20%). Median fractional allelic imbalance indexes did not disclose any statistically significant difference, being 0.69 in both components (range 0.44–0.73 in large-cell and 0.33–0.82 in small-cell components). At FISH analysis, the combined neuroendocrine carcinoma no. 4 showed the presence of both signals in the large-cell component, in agreement with an absence of allelic imbalance at PAR regions, whereas the presence of X and an absence of Y signals was found in the small-cell component, indicating a complete loss of Y chromosome. The combined tumor no. 5 showed the presence of X and an absence of Y signal, indicative of a loss of Y chromosome in both components.

(a) Frequency of allelic imbalances at 11 chromosomal regions in the two tumor-cell components of six combined neuroendocrine carcinomas of the lung. (b) Genetic concordance between components in each combined carcinoma investigated. (c) Frequency of allelic imbalances at 11 chromosomal regions in eight pure large-cell neuroendocrine carcinomas (LCNEC) and eight pure small-cell lung carcinomas (SCLC).
Genetic Concordance Between the Two Components of Combined Neuroendocrine Carcinomas
A high degree of genetic concordance between the two components was found in five out of six combined neuroendocrine carcinomas, ranging from 82 to 100% of all informative chromosomal regions investigated (Figure 2b; Table 2). Only tumor no. 4 showed a very low level of concordance (18%) between the two components. This case displayed some microsatellite instability in the large-cell component, although amplification of markers commonly used for instability testing (BAT26, BAT40 and L-MYC) failed to demonstrate a classic unstable pattern.
Allelic Imbalances in Pure Forms (n=8)
In both types of pure neoplasms, the frequency of allelic imbalance was found to be identical at region 5q (63% of cases, 5/8) and similar at 17p (83% in large-cell vs 88% in small-cell carcinomas) and 3p (75 vs 88%) (Figure 2c; Table 3). Allelic imbalances more frequently involving pure large-cell neuroendocrine carcinomas were found at 10q (75 vs 50%), X–Y PAR2 (75 vs 38%), X–Y PAR1 (57 vs 25%), 6p (50 vs 25%) and 9p (50 vs 38%). Allelic imbalances more frequently involving pure small-cell carcinomas were found at 16q (75 vs 38%), 4q (71 vs 43%) and 6q (25 vs 14%). Although remarkable at some chromosomal regions, none of these differences reached statistical significance. Median fractional allelic imbalance index was 0.60 in pure large-cell neuroendocrine (range 0.2–1.0) and 0.55 in pure small-cell carcinomas (range 0.09–0.80), a difference not significant.
FISH data showed the presence of orange signal for the CEP X probe and an absence of the green signal for the CEP Y probe, indicating a complete loss of Y chromosome in both cases of pure large-cell carcinomas from male patients with multiple imbalances at PAR regions (nos. 13 and 14). FISH data showed the presence of both X and Y signals in the two pure small-cell carcinomas from male patients with multiple imbalances at PAR regions (no. 21 and 22). Case no. 21 was characterized by an absence of Y signal in 30% of cells, whereas case no. 22 revealed a polysomic pattern, with an over-representation of both sex chromosomes (average number of signals per cell: 2.9 for X and 1.4 for Y). Thus, in this case, the X chromosome increased copy number most likely accounted for the allelic imbalances at PAR region observed at microallelotyping.
Comparison of Alterations in Combined and Pure Forms
The comparison of large-cell neuroendocrine components of combined neoplasms vs pure large-cell neuroendocrine carcinomas disclosed similar frequencies of allelic imbalance, with differences below 25%, at 3p, 5q, 9p, 10q, 17p and X–Y PAR1 regions. Wider differences, above 25%, were found at the following regions: 4q (83% combined vs 43% pure), 6p (20 vs 50%), 6q (80 vs 14%; P=0.072), 16q (83 vs 38%) and X–Y PAR2 (50 vs 75%). Only the differences at 6q regions approached statistical significance (Figure 2a and c).
The comparison of small-cell components of combined neoplasms vs pure small-cell carcinomas disclosed similar frequencies of allelic imbalance at 3p, 4q, 5q, 6p, 6q, 9p, 10q, 16q and 17p regions, with differences below 25%. Differences above 25%, although not statistically significant, were found only at X–Y PAR1 (50% combined vs 25% pure) and X–Y PAR2 (67 vs 38%).
Discussion
The combined small-cell/large-cell neuroendocrine carcinoma variant is a pulmonary neoplasm with biphenotypic neuroendocrine differentiation that has never been extensively studied. To our knowledge, this is the first study in which a separate genetic characterization of the two components of this combined neoplasm was performed. Ullmann et al6 performed a comparative genomic hybridization (CGH) study on five mixed small-cell/large-cell neuroendocrine carcinomas of the lung, but no discrimination between the two phenotypic components was reported. In the present investigation, only combined neoplasms with physically separated large-cell and small-cell areas were taken into account to allow separate microdissection, DNA extraction and genetic analysis through microallelotyping of the two components. Microallelotype analysis is a useful tool to disclose allelic imbalances, mostly reflecting the loss of tumor-suppressor genes, and to define the polyclonal or monoclonal origin of mixed, collision and multiple tumors, by comparison of the respective molecular patterns.14, 15, 16
In the present series of combined neuroendocrine neoplasms, a common background of allelic imbalances similarly involving both components was demonstrated. These genetic alterations mostly targeted chromosomal regions known to harbor tumor-suppressor genes frequently involved in lung carcinogenesis, namely 17p13.1, 3p14.2–3p21.2, 5q21 and 9p21.7, 17, 18, 19 Alterations of chromosomes 3p, 9p21 and 17p13 are frequently found even in precursor lesions and, therefore, are considered as critical steps in early tumorigenesis.17 The report of common genetic alterations, especially in chromosomal regions involved in early carcinogenesis, suggests a close genetic relationship between the two phenotypically different components of these combined neoplasms. When the genetic relationship between the distinct components of each biphenotypic neuroendocrine neoplasm was considered, more similarities emerged. In fact, in the majority of patients the two components showed a high degree of genetic concordance, represented by either lack of alterations or presence of imbalances involving the very same allele. These similarities support the hypothesis of a monoclonal carcinogenesis mechanism, with tumor cells of the two components deriving from a common precursor undergoing divergent differentiation.
In keeping with this interpretation is our observation of 4q12–4q24 imbalances occurring with the same high frequency (83%) in both components of our neuroendocrine combined cancers. On the contrary, the literature on pure forms has reported that losses at 4q21 and 4q24 occur exclusively in small-cell lung carcinomas, being observed in 40% of these, whereas absent in large-cell neuroendocrine carcinomas.7 Our data on pure neoplasms, although indicating a certain degree of 4q alterations also in large-cell carcinomas (43%), are in agreement with the literature, confirming their prevalence in small-cell lung carcinomas (71%). Therefore, the distinct neuroendocrine components of combined neoplasms appear to be more similar to each other than to their respective pure forms, another clue supporting the close genetic relationship between the two components.
Besides a common background of genetic alterations in both components, differences were also observed in combined neoplasms, suggesting divergent differentiation and paralleling morphological differences, even though no statistically significant differences were found, possibly due to the limited number of combined cases investigated.
The chromosomal regions more differentially altered in combined neoplasms were 6q, 10q and 16q in the large-cell component, and 6p and X–Y PAR regions in the small-cell component. Allelic losses on chromosome 10q are frequently reported in human neoplasms, and, in lung carcinomas, have been suggested as being associated with tumor progression and metastatic phenotype.7, 20 Recently, losses at 10q22.1–26.3 have been found to significantly correlate with poor prognosis in large-cell neuroendocrine carcinomas, thereby suggesting that tumor-suppressor gene(s) responsible for an aggressive phenotype may be harbored in that chromosomal region.7 In the present study, imbalances at 10q were more frequently observed in large-cell than in small-cell carcinomas, in both combined and pure tumors.
Recently, a high-resolution CGH analysis has reported losses at 16q21–24 as one of the newly identified chromosomal alterations common to both small and large-cell neuroendocrine carcinomas.7 Losses at 16q21–24 seem to be a specific neuroendocrine alteration, probably playing a key role in the pathogenesis of high-grade neuroendocrine tumors, being infrequently reported in non-small-cell lung carcinomas.7 In our study, the involvement of the 16q region in high-grade neuroendocrine tumors was confirmed. In combined neoplasms, allelic imbalances at 16q appeared to be slightly more frequent in the large- than in the small-cell component, whereas in pure forms this alteration was more prevalent in small-cell carcinomas.
Losses at 6q have frequently been reported in various human neoplasms. Among these, sporadic endocrine pancreatic tumors (sharing with neuroendocrine lung neoplasms a common origin from the embryonal foregut) present two hot spots at 6q22.1 and 6q23–q24, potentially harboring putative tumor-suppressor genes involved in their oncogenesis and malignant progression.21 In the present study, 6q imbalances had occurred in the large-cell component of combined neoplasms, with a frequency double that of the small-cell component (80 vs 40%). It is notable that this region was not substantially affected in the pure forms (below 30%), including the large-cell neuroendocrine carcinomas. Therefore, a marked difference in the frequency of 6q allelic imbalances was observed in pure large-cell neuroendocrine carcinomas vs the large-cell component of combined neoplasms, a difference approaching statistical significance despite the limited number of cases investigated.
In our combined neoplasms, imbalances at 6p25–21.3 occurred more frequently in the small- rather than in the large-cell component. Similar to region 4q, these data do not agree with those reported in the literature in the corresponding pure tumors. In fact, losses at 6p21.3, harboring the tumor necrosis factor-α, were reported more often in large- than in small-cell neuroendocrine carcinomas of the lung,7 a finding in agreement with our own data on pure forms, which show double the frequency of imbalances in pure large-cell- as compared with small-cell carcinomas (50 vs 25%).
A similar discrepancy between pure and combined high-grade neuroendocrine carcinomas of the lung was observed also at X–Y PAR regions, that is, those regions of sex chromosomes sharing X–Y homologies, pairing regularly at male meiosis and undergoing recombination.13 In previous studies by our laboratory, allelic losses on X chromosome were found to correlate with malignancy in foregut endocrine neoplasms11, 22 and colorectal cancers.13 In lung neuroendocrine tumors from female patients, X chromosome deletions proved to be a frequent event in large-cell carcinomas, but very rare in small-cell carcinomas.11 To verify this potentially discriminating marker also in male patients, we amplified microsatellite markers located in the X–Y PAR regions. Indeed, pure neuroendocrine lung neoplasms confirmed previous data,11 with a frequency of alterations higher in large- than in small-cell carcinomas. Conversely, in the two components of combined neoplasms, all cases showed a concordant genetic pattern, with the exception of case no. 4, which was characterized by allelic imbalances confined to the small-cell component alone. Interestingly, FISH analysis performed in male patients with imbalances at PAR regions demonstrated the complete absence of Y signal in pure large-cell carcinomas and both components of the combined tumors investigated. In these cases, therefore, Y chromosome appears to be the sex chromosome targeted by the deletions, suggesting the presence of putative tumor-suppressor gene(s) on it, as already suspected in sporadic colorectal cancers.13 Again, the mechanism of sex chromosome involvement would seem to be different in small-cell lung carcinomas, in which the reduction of Y signal is only relative, and in one case is due to over-representation of X signal rather than to Y allelic loss.
In conclusion, this is the first report, which describes a molecular characterization of the rare variant of small-cell lung carcinoma combined with large-cell neuroendocrine carcinoma. Overall, the phenotypically different components of these combined neoplasms appear to be closely related, and the high degree of concordance between them strongly suggests a common origin. The distinct components of these combined neoplasms appear to be more similar to each other than to their respective pure forms. On the other hand, when differences did emerge, they often seemed quite specific for this histological variant as compared with pure forms. All these findings support the view that combined small-cell/large-cell neuroendocrine carcinomas do not represent a mere combination of different neuroendocrine growth patterns, but instead constitute transition carcinomas in the spectrum of high-grade neuroendocrine pulmonary tumors, where the two extremes are represented by the corresponding pure forms.
References
Asamura H, Kameya T, Matsuno Y, et al. Neuroendocrine neoplasms of the lung: a prognostic spectrum. J Clin Oncol 2006;24:70–76.
Travis WD . The concept of pulmonary neuroendocrine tumours. In: Travis WD, Brambilla E, Müller-Hermelink HK, Harris CC (eds). World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart. IARC Press: Lyon, 2004, pp 19–20.
Travis WD, Linnoila RI, Tsokos MG, et al. Neuroendocrine tumors of the lung with proposed criteria for large-cell neuroendocrine carcinoma. An ultrastructural, immunohistochemical, and flow cytometric study of 35 cases. Am J Surg Pathol 1991;15:529–553.
Marchevsky AM, Gal AA, Shah S, et al. Morphometry confirms the presence of considerable nuclear size overlap between “small-cells” and “large-cells” in high-grade pulmonary neuroendocrine neoplasms. Am J Clin Pathol 2001;116:466–472.
Nicholson SA, Beasley MB, Brambilla E, et al. Small-cell lung carcinoma (SCLC): a clinicopathologic study of 100 cases with surgical specimens. Am J Surg Pathol 2002;26:1184–1197.
Ullmann R, Petzmann S, Sharma A, et al. Chromosomal aberrations in a series of large-cell neuroendocrine carcinomas: unexpected divergence from small-cell carcinoma of the lung. Hum Pathol 2001;32:1059–1063.
Peng WX, Shibata T, Katoh H, et al. Array-based comparative genomic hybridization analysis of high-grade neuroendocrine tumors of the lung. Cancer Sci 2005;96:661–667.
Jones MH, Virtanen C, Honjoh D, et al. Two prognostically significant subtypes of high-grade lung neuroendocrine tumours independent of small-cell and large-cell neuroendocrine carcinomas identified by gene expression profiles. Lancet 2004;363:775–781.
Travis WD, Brambilla E, Müller-Hermelink HK, et al (eds). World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart. IARC Press: Lyon, 2004.
Hiroshima K, Iyoda A, Shibuya K, et al. Genetic alterations in early-stage pulmonary large-cell neuroendocrine carcinoma. Cancer 2004;100:1190–1198.
D'Adda T, Bottarelli L, Azzoni C, et al. Malignancy-associated X chromosome allelic losses in foregut endocrine neoplasms: further evidence from lung tumors. Mod Pathol 2005;18:795–805.
Arvanitis DA, Angelakis E, Koumantakis EE, et al. Allelic imbalance in hMLH1 or BRCA2 loci associated with response of cervical and endometrial cancer to radiotherapy. Int J Mol Med 2002;10:55–63.
Bottarelli L, Azzoni C, Necchi F, et al. Sex chromosome alterations associate with tumor progression in sporadic colorectal carcinomas. Clin Cancer Res 2007;13:4365–4370.
Vortmeyer AO, Lubensky IA, Merino MJ, et al. Concordance of genetic alterations in poorly differentiated colorectal neuroendocrine carcinomas and associated adenocarcinomas. J Natl Cancer Inst 1997;89:1448–1453.
Huang J, Behrens C, Wistuba II, et al. Clonality of combined tumors. Arch Pathol Lab Med 2002;126:437–441.
Furlan D, Cerutti R, Genasetti A, et al. Microallelotyping defines the monoclonal or the polyclonal origin of mixed and collision endocrine-exocrine tumors of the gut. Lab Invest 2003;83:963–971.
Dacic S . Molecular profiling of lung carcinoma: identifying clinically useful tumor markers for diagnosis and prognosis. Expert Rev Mol Diagn 2007;7:77–86.
Kobayashi Y, Tokuchi Y, Hashimoto T, et al. Molecular markers for reinforcement of histological subclassification of neuroendocrine lung tumors. Cancer Sci 2004;95:334–341.
Onuki N, Wistuba II, Travis WD, et al. Genetic changes in the spectrum of neuroendocrine lung tumors. Cancer 1999;85:600–607.
Petersen S, Wolf G, Bockmühl U, et al. Allelic loss on chromosome 10q in human lung cancer: association with tumour progression and metastatic phenotype. Br J Cancer 1998;77:270–276.
Barghorn A, Speel EJM, Farspour B, et al. Putative tumor suppressor loci at 6q22 and 6q23–q24 are involved in the malignant progression of sporadic endocrine pancreatic tumors. Am J Pathol 2001;158:1903–1911.
Pizzi S, D'Adda T, Azzoni C, et al. Malignancy-associated allelic losses on the X-chromosome in foregut but not in midgut endocrine tumours. J Pathol 2002;196:401–407.
Acknowledgements
This work was supported by grants from the Italian Ministry of University and Research (MIUR-PRIN, grant number 20050692205_001 to CB) and the Italian Association for Cancer Research (AIRC to GP). We thank Professor Mauro Papotti, University of Turin, for providing case nos. 7, 8, 15 and 16; Professor Giulio Rossi, University of Modena, for providing case no. 20 and Mrs Emilia Corradini, University of Parma, for her excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of interest
None declared.
Rights and permissions
About this article
Cite this article
D'Adda, T., Pelosi, G., Lagrasta, C. et al. Genetic alterations in combined neuroendocrine neoplasms of the lung. Mod Pathol 21, 414–422 (2008). https://doi.org/10.1038/modpathol.3801014
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3801014
Keywords
This article is cited by
-
Clinical relevance of neuroendocrine differentiation in lung adenocarcinoma
The Chinese-German Journal of Clinical Oncology (2012)
-
Applying unmixing to gene expression data for tumor phylogeny inference
BMC Bioinformatics (2010)
-
Genetics of a combined lung small cell carcinoma and large cell neuroendocrine carcinoma with adenocarcinoma
Virchows Archiv (2008)
