
Abstract
Primary pulmonary and mediastinal synovial sarcoma is rare and poses a diagnostic challenge particularly when unusual histological features are present. We present 60 cases of primary pulmonary and mediastinal synovial sarcoma (29 male and 27 female subjects; mean age, 42 years) and compare our results with five prior series to better define unusual histological features. Clinically, patients with mediastinal synovial sarcoma were younger with a male gender bias. Radiologically, tumors were well delineated with distinctive magnetic resonance imaging features and little vascular enhancement. In all, 21/46 patients died of disease within 5 years. Histologically, all tumors had dense cellularity, interlacing fascicles, hyalinized stroma, and mast cell influx. Hemangiopericytoma-like vasculature (48/60), focal myxoid change (30/60), and entrapped pneumocytes (23/60) were seen. Calcification was not prevalent (10/60). Unusual histological features included Verocay body-like formations (7/60), vague rosettes (6/60), well-formed papillary structures (3/60), adenomatoid change (3/60), and rhabdoid morphology (2/60). Immunohistochemistry demonstrated expression of pancytokeratin (39/58), epithelial membrane antigen (29/53), cytokeratin 7 (26/40), cytokeratin 5/6 (5/7), calretinin (15/23), CD99 (19/23), bcl-2 (24/24), CD56 (11/11), S-100 (9/51), and smooth muscle actin (8/32). In total, 92% (36/39) of primary pulmonary and mediastinal synovial sarcomas studied were positive for t(x;18). In conclusion, our study confirms the clinical, histological, immunohistochemical, and molecular data from previous large series of primary pulmonary and mediastinal synovial sarcoma. Compared with soft tissue synovial sarcoma, primary pulmonary and mediastinal synovial sarcoma has less calcification, less obvious mast cell influx, and less radiologic vascularity, but similar magnetic resonance imaging features, percentage of poorly differentiated tumors, and number of t(x;18)-positive tumors. Awareness of focal unusual histology can prevent misdiagnosis particularly in t(x;18)-negative tumors.
Similar content being viewed by others

Main
Soft tissue synovial sarcoma is a clinicopathologically and cytogenetically distinctive neoplasm that mainly affects deep soft tissues of the extremities in adolescents and young adults.1 Synovial sarcoma is also recognized as a primary pulmonary and mediastinal neoplasm.2, 3 Primary pulmonary and mediastinal synovial sarcoma is an aggressive tumor sharing common histological features with soft tissue synovial sarcoma.3, 4, 5, 6, 7 Molecular testing for the pathognomonic t(x;18) chromosomal translocation has enabled diagnostic confirmation in approximately 90% of cases.8 In t(x;18)-negative cases, diagnosis must rely on histological and immunophenotypic features. The differential diagnosis of primary pulmonary and mediastinal synovial sarcoma is particularly challenging when histological features unusual to synovial sarcoma, but common to other neoplasms are focally present. We evaluated the clinical, radiological, and pathological findings in 60 cases of primary pulmonary and mediastinal synovial sarcoma and compared our results with five prior large series to better define the unusual histological features of this entity.
Materials and methods
In total, 119 cases diagnosed as synovial sarcoma or sarcoma, from 1981 to 2006, were retrieved from the Armed Forces Institute of Pathology's Registry of Pulmonary and Mediastinal Pathology. Fifty-nine cases that failed to meet the World Health Organization histologic and/or immunohistochemical criteria for synovial sarcoma were excluded including metastatic and chest wall soft tissue tumors. Chest radiographs, computed tomography, magnetic resonance imaging, or positron emission tomography/computed tomography studies were available in 13 cases. Follow-up data were obtained from patient records. Hematoxylin and eosin-stained sections were available for each case (range, 1–47; mean, 8). Tumors were subtyped as monophasic or biphasic according to World Health Organization criteria.2 The monophasic subtype was comprised solely of elongated spindle cells arranged in densely cellular interweaving fascicles. Biphasic tumors contained spindle as well as epithelial components; in the latter, cytologically malignant epithelial cells formed gland-like spaces with tubulo-papillary formations. Grading by tumor cell differentiation, mitotic rate, and necrosis was performed following the French Federation of Cancer Centers (FNCLCC) scheme. Histological features were categorized as major, minor, or unusual based on frequency and/or diagnostic utility. Immunohistochemistry was performed on paraffin-embedded sections using commercially available antibodies (Table 1). Molecular analysis was performed on RNA extracted from paraffin-embedded samples. SYT/SSX RNA fusion transcripts resulting from t(x;18)(p11;q11) translocation were detected using real-time reverse transcriptase-polymerase chain reaction.9 Subtyping of SYT/SSX 1 and 2 fusion transcripts was performed using methods previously described.9 In three cases, the molecular analysis was performed at outside laboratories.
Results
Clinical Findings
Pertinent clinical features are presented in Table 2. The study group included 29 male and 27 female subjects ranging from 10 to 84 years of age (mean, 42 years). The most common presenting symptoms were dyspnea and chest pain. Tumors were distributed in the lung (58%, 34/58), pleura (33%, 19/58), and mediastinum (9%, 5/58). Surgical resection of the primary tumor included pneumonectomy (16%, 9/55), lobectomy (40%, 22/55), and excision of the tumor mass (18%, 10/55). Patients developed local recurrence (18%, 7/38) within 6–49 months (mean, 17 months). Metastases in eight cases were to bilateral lungs, regional lymph nodes, abdomen, liver, and spleen, ranging from the time of diagnosis to 49 months following diagnosis. Forty-eight percent (22/46) of patients died of disease within 1–74 months (mean, 23) and 46% (21/46) died of disease within 5 years. Twenty-six percent (12/46) of patients were alive with no evidence of disease and 7% (3/46) were alive with disease at the time of this report. An additional 19% (9/46) were alive, but information on disease status was unavailable. Follow-up (46/60 cases) ranged from 2 to 249 months (mean, 37). No cases had clinical or radiological evidence of another primary tumor site.
Radiologic Findings
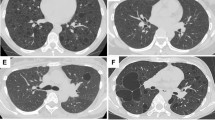
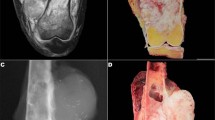
Radiology included 10 chest radiographs, 12 computed tomography studies, three magnetic resonance imaging studies, and one case with fluorine-18-fluorodeoxyglucose positron emission tomography/computed tomography images. On chest radiographs, tumors appeared homogeneous with sharply marginated borders (80%, 8/10) and contralateral mediastinal shift (10%, 1/10). There was no evidence of cavitation, calcification, or lymphadenopathy. On computed tomography, tumors were pleural based (75%, 9/12) and showed homogeneous or heterogeneous enhancement (Figure 1). Ipsilateral pleural effusion was common (67%, 8/12), while enhancing tumor vessels were rare (8%, 1/12). There was no evidence of bone destruction, chest wall invasion, or calcification. On magnetic resonance, T1-weighted and T2-weighted images showed a well-circumscribed mass without evidence of spiculation or satellite nodules (100%, 3/3). T2-weighted images included nodular areas of intermediate signal intensity combined with punctuate or cystic areas of high signal intensity suggestive of necrosis, hemorrhage, or myxoid material (67%, 2/3) (Figure 2). Following administration of gadopentetate dimeglumine, magnetic resonance imaging demonstrated primary pulmonary and mediastinal synovial sarcoma to have a peripheral rim of enhancement (100%, 3/3). Positron emission tomography/computed tomography images demonstrated focal increased uptake of fluorine-18-fluorodeoxyglucose by the tumor (100%, 1/1).

Synovial sarcoma in 37-year-old man with chest pain and right arm swelling. PA chest radiograph (a) coned to the upper chest demonstrates a right apical mass. Interface with the chest wall suggests an extrapulmonary origin. A pleural-based nodule (arrow) is inferior to the mass. Contrast-enhanced CT scan (b) demonstrates a predominantly cystic mass with an eccentrically thickened wall (asterisk) and a metastatic nodule (arrowhead) along the lateral pleural surface.

Synovial sarcoma in a 36-year-old male with dyspnea and pleuritic chest pain. PA chest radiograph (a) coned to the right lung demonstrates a right hilar mass with smooth borders. Contrast-enhanced CT scan (b) demonstrates a large low attenuation mass with rim enhancement (arrowhead) and internal septations. Axial T2-weighted (2110/57.6) magnetic resonance image (c) shows greater contrast among internal components, with well-demarcated spaces (arrowheads) which suggest cysts.
Gross and Histologic Findings
Gross and histological findings are presented in Table 3. Tumors ranged in size from 0.6 to 17 cm (mean, 7.5) and were well-circumscribed, soft, tan masses with foci of necrosis, hemorrhage, and cystic change. Histologically, tumors were monophasic (88%, 53/60) or biphasic (12%, 7/60). Mast cells were not readily identified in hematoxylin and eosin-stained sections but were obvious on CD117 (100%; 31/31). Tumors were grade 2 (75%, 45/60) and grade 3 (poorly differentiated, 25%, 15/60) according to FNCLCC grading. Among poorly differentiated tumors, 14 were monophasic and one was biphasic. Tumor cell morphology included spindle cells (58%, 35/60) or a combination of spindle and round/epithelioid cells (42%, 25/60). Mitoses ranged from 1 to 95 per 10 high power fields (mean, 14), and included 0–9 (56%, 34/60), 10–19 (22%, 13/60), and 20 or more (22%, 13/60) per 10 high power fields. Necrosis was present in most tumors (75%, 45/60) and extensive (50% or more) in a few (12%, 7/60).
Major histological features included dense cellularity (100%, 60/60), interlacing fascicles (100%, 60/60), hyalinized or eosinophilic stroma (100%, 60/60), hemangiopericytoma-like vasculature (80%, 48/60), and focal myxoid change (50%, 30/60) (Figure 3a–d). Benign entrapped pneumocytes were common (38%, 23/60) (Figure 3e and f). Minor histological features included well-demarcated tumor margins (89%, 40/45), calcification (17%, 10/60), and focal cystic change (13%, 8/60). Tumors had at least a partial rim of fibrous tissue that abutted bronchial cartilage or pleura (78%, 35/45). Unusual histological features were focal, noted in at least one but no more than four slides per case, varying from 4 to 90 high power fields and included Verocay body-like areas (12%, 7/60), vague rosette formation (10%, 6/60), well-formed papillary structures (5%, 3/60), adenomatoid areas (5%, 3/60), and rhabdoid morphology (3%, 2/60) (Figure 3h–k).

Histologic features of synovial sarcoma. Synovial sarcoma is one of the most cellular spindle cell neoplasms of the lung and mediastinum and is characteristically composed of densely cellular interlacing fascicles (a). Densely cellular areas may alternate with less cellular myxoid foci (b). Hyalinized stroma may be prominent and mimic that of solitary fibrous tumor (c). Tumor vasculature is typically hemangiopericytoma-like with staghorn morphology (d). Entrapped pulmonary epithelium (e) can readily be misinterpreted as the epithelial component of a biphasic tumor; thyroid transcription factor-1 immunoreactivity is helpful in avoiding this pitfall (f). Cytokeratin positivity is critical for diagnosis and can be minimal (g). Unusual features that may lead to misdiagnosis include papillary formations (h), Verocay body-like areas (i), adenomatoid appearance (j), and rhabdoid morphology (k). Poorly differentiated tumors are defined by French Federation of Cancer Centers (FNCLCC) criteria and include round cell morphology and high mitotic rate (l).
Immunohistochemical and Molecular Findings
Immunohistochemical findings are presented in Table 4 and molecular findings in Table 5. Immunohistochemical studies showed focal positive membranous or cytoplasmic staining for epithelial markers (Figure 3g) including pancytokeratin (67%, 39/58), epithelial membrane antigen (55%, 29/53), and cytokeratin 7 (65%, 26/40), while 41% (15/37) of cases showed focal immunoreactivity for all three epithelial markers. Tumors were also focally positive for cytokeratin 5/6 (71%, 5/7). Diffuse immunoreactivity was seen with bcl-2 (100%, 24/24) and CD99 (83%, 19/23). Focal immunoreactivity was present with CD56 (100%, 11/11), calretinin (65%, 15/23), S-100 (18%, 9/51), and smooth muscle actin (25%, 8/32). Entrapped benign pneumocytes, present in 23 cases, were immunoreactive with thyroid transcription factor-1 and epithelial markers (Figure 3e and f).
The chromosomal translocation t(x;18) was present in 92% (36/39) of cases studied in which 58% (19/33) were fusion type SYT/SSX1 and 42% (14/33) fusion type SYT/SSX2. In three cases, the molecular analysis was performed at outside laboratories. While these cases were reported as t(x;18) positive, information on the SSX fusion type was unavailable. A greater percentage of patients who died of their disease within 5 years had tumors with SYT/SSX1 (67%, 8/12) compared with SYT/SSX2 (33%, 4/12). The association between fusion types and other factors is presented in Table 6.
Discussion
Synovial sarcoma, although rare, is a primary pulmonary and mediastinal neoplasm with distinctive histology. The presence of focal, unusual histological findings characteristic of more common epithelial and mesenchymal tumors may lead to misdiagnosis. This is particularly problematic in small biopsies or in primary pulmonary and mediastinal synovial sarcomas that are negative for the pathognomonic t(x;18) translocation. We present 60 primary pulmonary and mediastinal synovial sarcoma cases to better define unusual histological features and compare our results with five prior series.
Results from five prior series3, 4, 5, 6, 7 indicate that, in contrast to soft tissue synovial sarcoma, primary pulmonary and mediastinal synovial sarcoma occurs in older patients without gender bias. Clinical symptoms are site specific with few asymptomatic cases.4 Most primary pulmonary and mediastinal synovial sarcomas are located in the lung parenchyma,3, 4, 5, 6, 7 and rarely extend into the bronchial tree5 or occur in the heart or pericardium.4, 10, 11 Based on 2-year local recurrence rates3, 5, 6, 7 and poor 5-year disease-specific survival rates,4 primary pulmonary and mediastinal synovial sarcoma is more aggressive than soft tissue synovial sarcoma. Histologically, primary pulmonary and mediastinal synovial sarcoma reportedly shares identical features with soft tissue synovial sarcoma including dense cellularity, interlacing fascicles, hyalinized stroma, hemangiopericytoma-like vasculature, focal myxoid change, and mast cell influx.3, 4, 5, 6, 7 The monophasic subtype is most common, and studies have pointed out that entrapped pneumocytes should not be mistaken for the epithelial component of a biphasic tumor. Begueret et al4 reported that almost 40% of primary pulmonary and mediastinal synovial sarcomas were poorly differentiated, a finding consistent with the tumors' aggressive behavior.4 With rare exceptions, primary pulmonary and mediastinal synovial sarcomas demonstrate focal immunoreactivity with at least one epithelial marker.4 Additionally, the majority of t(x;18)-positive primary pulmonary and mediastinal synovial sarcomas contain the SYT/SSX1 fusion transcript.4, 5, 6 The only large primary pulmonary and mediastinal synovial sarcoma study with molecular data found no relationship between SYT/SSX fusion type and outcome.4
In addition to confirming results from prior series, our study elucidated some novel findings. Radiologically, compared with soft tissue synovial sarcoma, primary pulmonary and mediastinal synovial sarcoma shows less vascularity and a similar ‘triple sign’ (bright, dark, and gray) representing tumor, hemorrhage, and necrosis on magnetic resonance imaging. Our radiologic images, documented in a recent review,12 support the diagnosis of primary pulmonary and mediastinal synovial sarcoma. On chest radiographs, the lesion is typically uniform with well-circumscribed rounded or lobulated borders13, 14, 15 with mediastinal shift in some patients.16 Computed tomography demonstrates a well-defined homogeneous or heterogeneously enhancing mass containing necrotic areas and soft tissue components (Figure 1). Ipsilateral pleural effusion is common,5, 13, 14, 15, 16 while mediastinal lymphadenopathy is rare.13 Sarcomas arising in the chest wall closely match the imaging characteristics of soft tissue synovial sarcoma with cortical bone destruction, tumor calcification and/or invasion into chest wall musculature,17, 18 features not observed on imaging in our study. Magnetic resonance imaging reveals more dramatic heterogeneity within tumors than demonstrated by computed tomography (Figure 2).13, 17 The T2-weighted images on magnetic resonance imaging in our series demonstrate the so-called ‘triple sign.’12 Soft tissue synovial sarcoma tends to be intermuscular and have vessels in one-third (representing the hemangiopericytoma-like pattern), which were only rarely observed on chest imaging in the current series.
While we found relatively less frequent intratumoral calcifications in primary pulmonary and mediastinal synovial sarcoma compared to soft tissue synovial sarcoma, our study found no diagnostically significant histological differences between these two tumors. However, the relatively large sample size helped identify focal unusual histology in primary pulmonary and mediastinal synovial sarcoma that can erroneously suggest more common primary and metastatic pulmonary neoplasms (Figure 3). Unusual histological features may be particularly challenging in small biopsies or when t(x;18) is negative. Verocay body-like areas can occur in primary pulmonary and mediastinal synovial sarcoma,3 which are similar to those seen in malignant peripheral nerve sheath tumor.10 The stromal background of malignant peripheral nerve sheath tumor, however, typically lacks hyalinization and appears more basophilic. While focal immunoreactivity for S-100 can be present in both tumors,10 primary pulmonary and mediastinal synovial sarcoma is often immunoreactive for cytokeratin 7, a finding not generally seen in malignant peripheral nerve sheath tumor.19 Clinically, malignant peripheral nerve sheath tumors arise from nerve or neurofibroma and are associated with neurofibromatosis type I in approximately two-thirds of cases.20
Primary pulmonary and mediastinal synovial sarcoma with focal vague rosette formation can lead to misdiagnosis as primitive neuroectodermal tumor.8, 10 Primary pulmonary and mediastinal synovial sarcoma may also be reminiscent of primitive neuroectodermal tumor when the former is poorly differentiated and displays round cell morphology. Unlike primary pulmonary and mediastinal synovial sarcoma, primitive neuroectodermal tumor typically has distinct cell borders, clear cytoplasm, scant stroma, and lacks hemangiopericytoma-like vasculature. Both tumors can express CD99, CD56, and cytokeratins,10, 21 although expression of cytokeratin 7 makes a diagnosis of primitive neuroectodermal tumor less likely.21 Chromosomal translocation t(11;22) is present in 85% of primitive neuroectodermal tumors.8
Focal well-formed papillary7 or adenomatoid areas in primary pulmonary and mediastinal synovial sarcoma may be misinterpreted as carcinoma or malignant mesothelioma. In particular, pleomorphic carcinoma can present with spindle cell and adenocarcinoma components. Carcinomas are more cytologically atypical with greater pleomorphism than primary pulmonary and mediastinal synovial sarcoma. Carcinomas may have areas of squamous differentiation or contain tumor giant cells, features not observed in primary pulmonary and mediastinal synovial sarcoma. While cytokeratins are focally expressed in primary pulmonary and mediastinal synovial sarcoma, diffuse positivity for epithelial markers is not characteristic. However, it should be kept in mind that spindle cell carcinomas may also be only focally positive for cytokeratins. Carcinomas are more likely to show an infiltrative growth pattern, regional lymph node involvement, and/or widespread metastases. Radiological correlation can be useful in this setting as malignant mesothelioma typically presents with diffuse pleural involvement rather than forming a solitary pulmonary mass. Although rare, localized malignant mesothelioma may present as a pleural mass that extends into the pulmonary parenchyma. Our results demonstrate that tumor cells in primary pulmonary and mediastinal synovial sarcoma may be focally positive for calretinin and CK5/6, thus, in the most challenging cases, molecular testing for t(x;18) may be helpful.
Hypercellular areas in solitary fibrous tumor can pose a diagnostic challenge particularly in small biopsies. Similar to primary pulmonary and mediastinal synovial sarcoma, solitary fibrous tumor demonstrates a collagenous stroma, hemangiopericytoma-like vasculature, and sometimes myxoid change. However, solitary fibrous tumor overall is less densely cellular and characteristically has a pronounced mixture of hypocellular and hypercellular areas. The collagenous stroma of primary pulmonary and mediastinal synovial sarcoma is generally more focal and less densely hyalinized than the relatively diffuse ‘ropy’ collagen associated with solitary fibrous tumor. Primary pulmonary and mediastinal synovial sarcoma is immunoreactive with epithelial markers and is negative for CD34, while solitary fibrous tumor is negative with epithelial markers and typically demonstrates diffuse CD34 positivity.2 Bcl-2 is not discriminatory as it is diffusely positive in both tumors. When present, the clinical features of hypertrophic osteoarthropathy, symptomatic hypoglycemia due to secretion of an insulin-like growth factor, and attachment to the visceral pleura by a pedicle support the diagnosis of solitary fibrous tumor.2
Our series included 25% poorly differentiated tumors, which is similar to that observed in soft tissue synovial sarcoma (Figure 3l).1 The difference in number of poorly differentiated tumors between our study and Begueret et al4 may be partially explained by the fact that their study population had a greater mean tumor size and higher percentage of tumors larger than 5 cm. As Begueret et al4 point out, interobserver variability may contribute to overestimation of poorly differentiated tumors. Interobserver variability in application of tumor grading criteria can be virtually eliminated by use of the reproducible FNCLCC grading scheme, which has prognostic value.22
Errors in the histological subtyping of primary pulmonary and mediastinal synovial sarcoma can occur when entrapped pneumocytes are misinterpreted as the epithelial component of biphasic tumors. Entrapped pneumocytes are cytologically benign, are typically arranged in small clusters and/or well-formed gland-like structures at the periphery of the tumor (Figure 3e), and are immunoreactive with thyroid transcription factor-1 (Figure 3f). In contrast, the epithelial component of biphasic primary pulmonary and mediastinal synovial sarcoma is cytologically malignant, forms cleft-like spaces with tubulo-papillary features, and is negative for thyroid transcription factor-1.
Our results showed the majority of patients who died of disease within 5 years had tumors of the SYT/SSX1 fusion type. While several studies of soft tissue synovial sarcoma have suggested that the SYT/SSX fusion type is of prognostic value,23, 24, 25, 26 a more recent study has found no prognostic difference between fusion types.27 Our cases showed similar mean tumor size in both SYT/SSX1- and SYT/SSX2-positive tumors, and, interestingly, SYT/SSX1 had fewer FNCLCC grade 3 than grade 2 tumors. Patients with SYT/SSX1-positive tumors also had a younger mean age of 43 years compared with a mean of 54 years in patients with SYT/SSX2-positive tumors. It may be that primary pulmonary and mediastinal synovial sarcoma with the fusion type SYT/SSX1 affects younger patients and inherently carries a worse prognosis. Certainly, fusion products generated from chromosomal translocations have been shown to have meaningful clinical correlates in Ewing sarcoma/primitive neuroectodermal tumor (EWS-FLI1),28 alveolar rhabdomyosarcoma (PAX-FKHR),29 and chronic myelogenous and acute lymphoblastic leukemias (BCR-ABL).30
Our clinicopathological analysis of 60 primary pulmonary and mediastinal synovial sarcoma cases confirmed that this tumor occurs in older patients without gender bias. However, when evaluated independently, primary mediastinal synovial sarcoma occurs in younger patients with male predominance. Forty-six percent of our patients died of disease within 5 years after diagnosis, vs 35% for soft tissue synovial sarcoma1 confirming that primary pulmonary and mediastinal synovial sarcoma is more aggressive. Compared with soft tissue synovial sarcoma, primary pulmonary and mediastinal synovial sarcoma has less radiologic vascularity, but similar magnetic resonance imaging features. Histologically, primary pulmonary and mediastinal synovial sarcoma demonstrated features similar to soft tissue synovial sarcoma, including percentage of poorly differentiated tumors, but had less calcification and less obvious mast cell influx. The biphasic subtype was uncommon, in contrast to soft tissue synovial sarcoma in which a third or more are biphasic.10 Most primary pulmonary and mediastinal synovial sarcomas demonstrated focal immunoreactivity with at least one epithelial marker. Based on our molecular data, primary pulmonary and mediastinal synovial sarcoma can be expected to be negative for t(x;18) by polymerase chain reaction in approximately 10% of cases. In addition, the majority of our t(x;18)-positive cases contained the SYT/SSX1 fusion transcript. Attention to the major histological features of primary pulmonary and mediastinal synovial sarcoma and awareness of focal, unusual histology will prevent misdiagnosis in t(x;18)-negative tumors. Future studies are needed to better establish the relationship between fusion type and outcome in primary pulmonary and mediastinal synovial sarcoma. Additionally, they may help determine whether primary mediastinal synovial sarcoma has different clinical or biologic characteristics than its pulmonary or pleural counterparts.
References
Weiss SW, Goldblum JR (eds). Enzinger and Weiss's Soft Tissue Tumors, 4th edn. Mosby: St Louis, MO, 2001, pp 1502–1504.
Travis WD, Brambilla E, Muller-Hermelink HK, et al. World Health Organization Classification of Tumors. Pathology and Genetics of Tumors of the Lung, Pleura, Thymus, and Heart. IARC Press: Lyon, France, 2004, p 344.
Zeren H, Moran CA, Suster S, et al. Primary pulmonary sarcomas with features of monophasic synovial sarcoma: a clinicopathological, immunohistochemical, and ultrastructural study of 25 cases. Hum Pathol 1995;26:474–480.
Begueret H, Galateau-Salle F, Guillou L, et al. Primary intrathoracic synovial sarcoma: a clinicopathologic study of 40 t(x;18)-positive cases from the French Sarcoma Group and the Mesopath Group. Am J Surg Pathol 2005;29:339–346.
Essary LR, Vargas SO, Fletcher CD . Primary pleuropulmonary synovial sarcoma: reappraisal of a recently described anatomic subset. Cancer 2002;94:459–469.
Okamoto S, Hisaoka M, Daa T, et al. Primary pulmonary synovial sarcoma: a clinicopathologic, immunohistochemical, and molecular study of 11 cases. Hum Pathol 2004;35:850–856.
Suster S, Moran CA . Primary synovial sarcomas of the mediastinum: a clinicopathologic, immunohistochemical, and ultrastructural study of 15 cases. Am J Surg Pathol 2005;29:569–578.
Fletcher CD, Unni KK, Mertens FE . World Health Organization Classification of Tumors. Pathology and Genetics of Tumors of Soft Tissue and Bone. IARC Press: Lyon, France, 2002, p 427.
Bijwaard KE, Fetsch JF, Przygodzki R, et al. Detection of SYT-SSX fusion transcripts in archival synovial sarcomas by real-time reverse transcriptase-polymerase chain reaction. J Mol Diagn 2002;4:59–64.
Miettinen ME . Diagnostic Soft Tissue Pathology. Churchill Livingstone: New York, 2003, pp 463–468.
Nicholson AG, Rigby M, Lincoln C, et al. Synovial sarcoma of the heart. Histopathology 1997;30:349–352.
Frazier AA, Franks TJ, Pugatch RD, et al. From the archives of the AFIP: pleuropulmonary synovial sarcoma. Radiographics 2006;26:923–940.
Duran-Mendicuti A, Costello P, Vargas SO . Primary synovial sarcoma of the chest: radiographic and clinicopathologic correlation. J Thorac Imaging 2003;18:87–93.
Nicholson AG, Goldstraw P, Fisher C . Synovial sarcoma of the pleura and its differentiation from other primary pleural tumours: a clinicopathological and immunohistochemical review of three cases. Histopathology 1998;33:508–513.
Zaring RA, Roepke JE . Pathologic quiz case. Pulmonary mass in a patient presenting with a hemothorax. Diagnosis: primary pulmonary biphasic synovial sarcoma. Arch Pathol Lab Med 1999;123:1287–1289.
Gaertner E, Zeren EH, Fleming MV, et al. Biphasic synovial sarcomas arising in the pleural cavity. A clinicopathologic study of five cases. Am J Surg Pathol 1996;20:36–45.
Fujimoto K, Hashimoto S, Abe T, et al. Synovial sarcoma arising from the chest wall: MR imaging findings. Radiat Med 1997;15:411–414.
Murphey MD, Jelinek JS, Kransdorf MJ, et al. Imaging of synovial sarcoma (abstract). Radiology 1998;209(P):421.
Smith TA, Machen SK, Fisher C, et al. Usefulness of cytokeratin subsets for distinguishing monophasic synovial sarcoma from malignant peripheral nerve sheath tumor. Am J Clin Pathol 1999;112:641–648.
Kleihues P, Cavenee WK . World Health Organization Classification of Tumors. Pathology and Genetics of Tumors of the Nervous System. IARC Press: Lyon, France, 2000, pp 172–174.
Machen SK, Fisher C, Gautam RS, et al. Utility of cytokeratin subsets for distinguishing poorly differentiated synovial sarcoma from peripheral primitive neuroectodermal tumour. Histopathology 1998;33:501–507.
Hasegawa T, Yamamoto S, Yokoyama R, et al. Prognostic significance of grading and staging systems using MIB-1 score in adult patients with soft tissue sarcoma of the extremities and trunk. Cancer 2002;95:843–851.
Kawai A, Woodruff J, Healey JH, et al. SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma. N Engl J Med 1998;338:153–160.
Ladanyi M, Antonescu CR, Leung DH, et al. Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: a multi-institutional retrospective study of 243 patients. Cancer Res 2002;62:135–140.
Nilsson G, Skytting B, Xie Y, et al. The SYT-SSX1 variant of synovial sarcoma is associated with a high rate of tumor cell proliferation and poor clinical outcome. Cancer Res 1999;59:3180–3184.
Panagopoulos I, Mertens F, Isaksson M, et al. Clinical impact of molecular and cytogenetic findings in synovial sarcoma. Genes Chromosomes Cancer 2001;31:362–372.
Guillou L, Benhattar J, Bonichon F, et al. Histologic grade, but not SYT-SSX fusion type, is an important prognostic factor in patients with synovial sarcoma: a multicenter, retrospective analysis. J Clin Oncol 2004;22:4040–4050.
De Alava E, Kawai A, Healey JH, et al. EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing's sarcoma. J Clin Oncol 1998;16:1248–1255.
Kelly KM, Womer RB, Sorensen PH, et al. Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J Clin Oncol 1997;15:1831–1836.
Melo JV . The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood 1996;88:2375–2384.
Author information
Authors and Affiliations
Corresponding author
Additional information
The opinions and assertions contained herein are the expressed views of the authors and are not to be construed as official or reflecting the views of the Departments of the Army or Defense. This is a US Government work, and as such is in the public domain in the United States of America.
Disclosure/conflict of interest
None of the authors have any disclosures or conflicts of interest.
Rights and permissions
About this article
Cite this article
Hartel, P., Fanburg-Smith, J., Frazier, A. et al. Primary pulmonary and mediastinal synovial sarcoma: a clinicopathologic study of 60 cases and comparison with five prior series. Mod Pathol 20, 760–769 (2007). https://doi.org/10.1038/modpathol.3800795
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800795
Keywords
This article is cited by
-
Usefulness of SS18-SSX antibody as a diagnostic marker for pulmonary metastatic synovial sarcoma
Diagnostic Pathology (2021)
-
Primary synovial sarcoma of the mediastinum: a rare tumor diagnosed by endoscopic ultrasound-fine needle biopsy (EUS-FNB)—Cytomorphologic, immunohistochemical, and molecular analysis
Clinical Journal of Gastroenterology (2021)
-
BRAF V600E mutation is a potential therapeutic target for a small subset of synovial sarcoma
Modern Pathology (2020)
-
Primary pleuropulmonary and mediastinal synovial sarcoma: a clinicopathologic and molecular study of 26 genetically confirmed cases in the largest institution of southwest China
Diagnostic Pathology (2016)
