
Abstract
Certain recurrent cytogenetic abnormalities are diagnostic of a specific neoplasm and may portend prognosis. As conventional cytogenetics may not reveal a neoplastic clone, and unfixed material for fluorescence in situ hybridization may be unavailable, performing fluorescence in situ hybridization on fixed tissues is diagnostically and prognostically valuable. Manual interpretation of fluorescence in situ hybridization signals may be difficult on paraffin-embedded tissue sections due to truncated nuclei. Therefore, we investigated the use of an automated image acquisition and analysis system (MetaSystems™) for interpretation of fluorescence in situ hybridization signals in tissue sections from dual fusion translocation probes. Three probe sets were analyzed on archival specimens with a confirmed diagnosis of mantle cell lymphoma, follicular lymphoma or Burkitt lymphoma. 100% of mantle cell lymphomas (7/7) were positive for t(11;14), 91% of follicular lymphomas (10/11) for t(14;18) and 100% of Burkitt lymphomas (9/9) for t(8;14). Successful hybridization was achieved using various tissue fixatives and fluorescence in situ hybridization interpretation was blinded with respect to the underlying diagnosis. Based on these results, automated analysis of fluorescence in situ hybridization on fixed tissues is accurate and valuable in the evaluation of B-cell lymphoma, and may provide pertinent diagnostic and prognostic information.
Similar content being viewed by others

Main
It is well known that certain recurrent cytogenetic abnormalities are the hallmark of neoplastic disease, including some subtypes of mature B-cell lymphomas. The presence of a particular abnormality may have diagnostic significance, such as t(14;18)(q32;q21) in follicular lymphoma, or prognostic/diagnostic significance such as t(11;14)(q13;q32) in mantle cell lymphoma.1 The ability to detect such chromosomal translocations is thus of utmost importance in the evaluation of clinical specimens.
The two cytogenetic methods that are utilized to determine the presence of chromosomal abnormalities include conventional cytogenetic studies and fluorescence in situ hybridization. Polymerase chain reaction DNA-based molecular studies may also be performed, but evaluation for particular translocations is often limited in paraffin-embedded, fixed tissues.2 For example, detection of the IgH/bcl-1 product in mantle cell lymphoma is reported to be in the range of 50%.3, 4 This necessarily implies that approximately 50% of cases may be falsely negative. Fluorescence in situ hybridization, on the other hand, has a detection sensitivity of essentially 100%,5 and is therefore usually the preferred testing modality.
As conventional cytogenetic and fluorescence in situ hybridization techniques require fresh, unfixed specimens, touch preparations or air-dried smears, and analysis is limited by the availability of these tissue types, the ability to assess the genetic aberrations on fixed, paraffin-embedded specimens has great potential in lymphoma diagnosis.
Recently, Paternoster et al6 described a successful method to extract nuclei from fixed tissues and perform fluorescence in situ hybridization on isolated, intact interphase nuclei. This method is straightforward and efficient. However, the nuclei extraction procedure requires preparation time for which smaller laboratories may not have technologist time available. In addition, the procedure disrupts tissue architecture and may intermix and dilute neoplastic cells with normal cells, skewing results.
As an alternative, we wanted to determine if fluorescence in situ hybridization analysis could be performed on whole sections of fixed, paraffin-embedded tissues and be generalized for use in the testing of routine clinical specimens for specific translocations, using dual fusion probes. Importantly, since tissue sectioning results in the truncation of nuclei (eg incomplete nuclear content), this method is not overtly amenable to the traditional method of manual scoring of numbers of signals (fusion or individual) per cell. Therefore, an alternative method, using an automated image acquisition and analysis system (eg MetaSystems™ (Altlussheim, Germany)) is preferable. This system uses a signal enumeration approach that compensates for the issues of incomplete nuclei,7 by placing tiles over the sample areas to approximate the area of a typical nuclear section. This approach, when validated as described here, is capable of determining in an accurate and unbiased manner the presence of an overall decrease or increase in the number of individual fluorescent signals or fusions per tumor cell (compared to non-tumor). Additionally, ‘virtual microdissections’ can be performed to target the analysis to the areas of interest, which are architecturally preserved on the tissue section, in order to ensure predominantly tumor cells are included in the final results. Fluorescence in situ hybridization analysis of translocations on paraffin-embedded whole tissue sections has been recently reported by several investigators,8, 9, 10, 11 but, to the best of our knowledge, an automated approach to the analysis has not been utilized.
In this context, we evaluated three commercially available translocation fluorescence in situ hybridization probes, on whole paraffin sections, which are currently used in the day-to-day evaluation of certain B-cell lymphomas and have diagnostic and prognostic significance. These probes (which were initially developed for use on cell suspensions, such as bone marrow aspirates) utilize the dual fusion strategy, and included t(11;14) for mantle cell lymphoma, t(14;18) for follicular lymphoma and t(8;14) for Burkitt lymphoma.
Materials and methods
Case Selection
Paraffin-embedded tissue blocks from 11 cases of Burkitt lymphoma, 13 cases of follicular lymphoma and seven cases of mantle cell lymphoma were identified from the pathology files at the University of New Mexico. A total of 12, 13 and 12 blocks were hybridized, respectively. Table 1 depicts characteristics of the archival tissue specimens that were selected for this study. All lymphoma diagnoses were verified by evaluation of the morphology (H&E), immunohistochemical, flow cytometric, and cytogenetic (when available) results. Cases of Burkitt lymphoma with a variant MYC translocation partner (eg t(2;8) or t(8;22)) (determined by conventional karyotyping), which lack fusion signals, were hybridized but ultimately excluded from this analysis, as we are reporting the ability to detect fusion signals in this study. Twenty paraffin-embedded tissue blocks from non-neoplastic lymph nodes were used as negative controls and to determine normal reference ranges for signal patterns.
Fluorescence In Situ Hybridization Protocol
Whole tissue sections (4 μm thick, unstained) were hybridized with LSI IGH/CCND1 dual color, dual fusion, LSI IGH/BCL-2 dual color, dual fusion, and LSI IGH/MYC, CEP8, tricolor, dual fusion translocation probe sets (Vysis™ Inc., Des Plaines, IL, USA). These probe sets correspond to the translocations, t(11;14)(q13;q32), t(14;18)(q32;q21) and t(8;14)(q24;q32) seen in mantle cell lymphoma, follicular lymphoma and Burkitt lymphoma, respectively. Each probe was hybridized to the 20 non-neoplastic tissues as well as their respective test lymphomas.
The sections were pretreated using a Vysis VP-2000 tissue processor (Vysis™, Inc., Des Plaines, IL, USA). Briefly this involves removing the paraffin with CitriSolv (Fisher Scientific, Fair Lawn, NJ, USA), treating the sections for 30 min in 1 M sodium thiocyanate, digesting for 60–90 min with protease at 0.5 mg/ml in 0.01 N HCl, and fixing in 10% neutral buffered formalin. Labeled probe (10 μl) are then applied to each tissue section, covered with a 22 mm2 coverslip, and sealed with rubber cement. The slides are placed on a Vysis HYBrite™ denaturation/hybridization system (Vysis™ Inc., Des Plaines, IL, USA), co-denatured with probe at 73°C for 6 min and allowed to hybridize at 37°C for 16–20 h. Hybridized slides are then washed in 2 × SSC containing 0.3% IGEPAL (Sigma-Aldrich, St Louis, MO, USA) at 73°C for 2 min, allowed to air dry and counterstained with 4′-6-Diamidino-2-phenylindole (DAPI).
Fluorescence In Situ Hybridization Analysis
The analysis was performed using the MetaSystems™ automated image acquisition and analysis system (Altlussheim, Germany). Areas involved by tumor (determined by comparing the nuclear DAPI appearance with the H&E section) were manually selected for image capture. ‘Regions of interest’ were selected within each captured image in order to ensure that only tumor cells were included in the analysis. The fluorescent signals from each ‘region of interest’ were then examined and analyzed on the MetaSystems™ Metafer 4 image analysis workstation.
Fluorescence in situ hybridization signals were enumerated by MetaSystems™ software, which provides a range of parameter adjustments for design of signal ‘classifiers’,7 and subsequently validated by the investigators (BH/AC/KR). The latter action is important in that the investigator essentially verifies the resultant data on digital images rather than through a microscope (as is done with conventional interphase fluorescence in situ hybridization on intact, individual cells). All three fluorescence in situ hybridization interpreters independently arrived at the final conclusion while blinded to the sample type. Unacceptable cells (due to poor morphology and/or fluorescence in situ hybridization signal quality) were deleted from the analysis. Although the time required for this activity is variable, depending on the results of the case and on the experience of the interpreter, it comprised approximately 15–30 min per case.
We required at least 1000 cells/‘tiles’ (see below) to be analyzed per case. The computer tabulates the results for each probe in each case, which are then displayed in histogram format with the x-axis representing the number of fusions detected starting at 0, 1, 2, etc. and the y-axis representing the total number of ‘tiles’ or ‘nuclear sections’ that contained certain numbers of fusions (linear). The histograms from neoplastic cases were compared with their respective non-neoplastic controls to assess for the presence or absence of increased fusion signals. Cases were considered positive when the peak fusion pattern shifted from a peak threshold of ‘0’ (as in the normal controls) to ‘1’ and ‘2’, thus indicating increased numbers of fusions per nucleus.
An important component of the classifier is setting an appropriate ‘tile size’ to a size roughly equivalent to the average diameter of a tumor cell. Tiling is a fluorescence in situ hybridization analysis automation method that allows detection of signals, and computation of distance between signals from different genes in areas of a tissue section with non-overlapping nuclei. The area, or tile size, is set to match the diameter of the involved nuclei in the tissue sample7 in order to calculate the number of signals and/or fusions per nuclear section. In this study, we were interested in assessing the number of fusion signals per nucleus. The number of fluorescence in situ hybridization signal fusion events (calculated as the number of translocation gene signal pairs with a separation distance less than a set value) per tile is a good approximation of the number of fusion events per nuclear section. This approach provides measurement of fusion events compared to normal control sections.
Another important role for the classifier is setting the value for the separation distance between probes, which when equal to or below that value, is considered a fusion signal. Normal separation distance values for non-fused signals for each fusion gene probe system (t(11;14), t(14;18), t(8;14)) were determined from tiling analysis of 20 normal lymph node tissue samples. This distance was generally in the range of 0.8–1.2 μm, with smaller tumor cell size correlating to smaller separation distances.
Classifier parameters used in this study can be obtained by contacting the authors.
Results
Characteristics of the Fluorescence In Situ Hybridization Technique on Fixed, Paraffin-Embedded Whole Tissue Sections
We achieved an overall fluorescence in situ hybridization probe hybridization success rate of >95% for all specimens, fixatives and probes combined. Importantly, this method does not require any additional tissue manipulation or cellular preparation time beyond that needed for cutting an unstained 4–5 μm thick section. In addition, it leaves the architecture of the fixed tissue intact with minimal alteration of the histologic arrangement. This allows for specific localization of the cells of interest and comparison (as needed) with the surrounding non-neoplastic cellular milieu.
We were able to achieve interpretable hybridization signals on a variety of tissue fixatives (formalin, B5, acetic acid zinc formalin and decalcified bone marrow core biopsies) and on archival tissues up to 7 years old. For the lymphoma specimens, the overall hybridization success rate for formalin-fixed tissues was 95% (20/21), B5-fixed tissues 57% (4/7), acetic acid zinc formalin-fixed tissues 100% (5/5) and decalcified bone marrow core biopsies 50% (2/4) (Table 2). For the non-neoplastic specimens, the overall hybridization success rate for formalin-fixed tissues was 100% (48/48) and for acetic acid zinc formalin-fixed tissues 100% (12/12) (Table 2). (Each set of 20 non-neoplastic tissues was hybridized with each of the three translocation probes).
Unsuccessful B5-fixed tissues tended to exhibit poor cellular morphology, weak/absent fluorescence in situ hybridization signals and high autofluorescence. Although only four cases were tested, bone marrow core biopsies were troublesome in that a portion/all of the tissue became detached from the slide. Obviously preservation of tissue and adequate morphology are critical for interpretation as one must be able to definitively ascertain if the cells being analyzed are truly tumor cells or are falsely negative because they represent the surrounding non-neoplastic background.
Automated Analysis
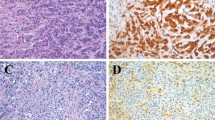
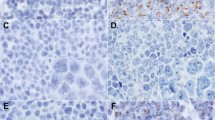
Upon completion of MetaSystems™ automated analysis, the frequency of fusion signals per nuclear section was displayed in histogram format. Based on the probe strategy used herein, cases without increased abnormal dual fusions (eg normal for that probe set) reveal a peak/majority of nuclei at ‘0’ (Figures 1a and 2a), whereas cases with increased fusions would reveal a peak at ‘1’ and ‘2’ (Figures 1b and 2b). It should be noted that normal cases will exhibit occasional random overlap of the green and red signals resulting in an artifactual single fusion; hence, the small number of nuclei in the histogram displaying a single fusion. The few nuclei in normal cases that show dual fusion signals represent the rare overlap of two different nuclei contained in the same ‘tile’ (enumerated by the computer as a single nucleus) which both have a single fusion in their respective nuclear section (Figure 1a).

(a) Formalin-fixed, tissue section of non-neoplastic lymph node hybridized with the Vysis™ LSI IGH/CCND1, dual color, dual fusion fluorescence in situ hybridization probe for the translocation t(11;14)(q13;q32). Normal fluorescence in situ hybridization pattern with two individual green (IgH) and orange (CCND1) signals per nuclear section (left panel). Higher magnification of representative fluorescence in situ hybridization image (upper right panel). Results from the automated analysis for fluorescence in situ hybridization fusion signals presented in histogram format (lower right panel). The x-axis represents the number of fusions per nuclear section, and the y-axis represents the total number of nuclear sections with that signal pattern (linear scale). The distribution of fusions in normal lymph node is centered on ‘0’ as expected. (b) Acetic acid zinc formalin-fixed, tissue section of mantle cell lymphoma in a lymph node hybridized with the Vysis™ LSI IGH/CCND1, dual color, dual fusion fluorescence in situ hybridization probe for the translocation t(11;14)(q13;q32). Abnormal positive fluorescence in situ hybridization pattern with increased numbers of yellow (fusion) signals per nuclear section (left panel). Higher magnification of representative fluorescence in situ hybridization image (upper right panel). Results from the automated analysis for fluorescence in situ hybridization fusion signals presented in histogram format (lower right panel). The x-axis represents the number of fusions per nuclear section, and the y-axis represents the total number of nuclear sections with that signal pattern (linear scale). The distribution of fusions in mantle cell lymphoma is centered on ‘1’ and ‘2’ as expected. The minor peak, which is also seen at ‘0’ most likely represents normal T-cells which are intimately admixed with the tumor cells.

(a) Formalin-fixed, tissue section of non-neoplastic lymph node hybridized with the Vysis™ LSI IGH/MYC, CEP8, tricolor, dual fusion fluorescence in situ hybridization probe for the translocation t(8;14)(q24;q32). Normal fluorescence in situ hybridization pattern with two individual green (IgH) and orange (MYC) signals per nuclear section (aqua not shown) (left panel). Higher magnification of representative fluorescence in situ hybridization image (upper right panel). Results from the automated analysis for fluorescence in situ hybridization fusion signals presented in histogram format (lower right panel). The x-axis represents the number of fusions per nuclear section, and the y-axis represents the total number of nuclear sections with that signal pattern (linear scale). The distribution of fusions in normal lymph node is centered on ‘0’ as expected. (b) Decalcified, bone marrow trephine biopsy section with Burkitt lymphoma hybridized with the Vysis™ LSI IGH/MYC, CEP8, tricolor, dual fusion fluorescence in situ hybridization probe for the translocation t(8;14)(q24;q32). Abnormal positive fluorescence in situ hybridization pattern with increased numbers of yellow (fusion) signals per nuclear section (aqua not shown) (left panel). Higher magnification of representative fluorescence in situ hybridization image (upper right panel). Results from the automated analysis for fluorescence in situ hybridization fusion signals presented in histogram format (lower right panel). The x-axis represents the number of fusions per nuclear section, and the y-axis represents the total number of nuclear sections with that signal pattern (linear scale). The distribution of fusions in Burkitt lymphoma is centered on ‘1’ and ‘2’ as expected.
Likewise, cases with an increased number of fusions (eg positive for that probe set), in addition to showing expected increased numbers of nuclei with increased fusions, will often also show a persistence (variable) of nuclear sections with the ‘normal’ ‘0’ pattern. This finding is attributable to the fact that background T-cells and other potential non-neoplastic, similarly-sized cells are intimately admixed with the tumor cells and are also being analyzed.
The range of single fusion signals, as compiled from the 20 non-neoplastic lymph node specimens, was 13–34% (mean 22%, median 24%) for the IgH/bcl-2 probe, 23–37% (mean 30%, median 29%) for the IgH/CCND1 probe and 11–35% (mean 29%, median 30%) for the IgH/MYC probe. The range of dual fusion signals was 1–5% (mean 3%, median 2%) for the IgH/bcl-2 probe, 2–7% (mean 4%, median 3%) for the IgH/CCND1 probe and 2–7% (mean 4%, median 4%) for the IgH/MYC probe.
The range of dual fusion signals (typical abnormal pattern), as compiled from the different types of lymphomas, was 12–31% (mean 24%, median 26%) for the IgH/bcl-2 probe, 8–30% (mean 22%, median 21%) for the IgH/CCND1 probe and 18–44% (mean 34%, median 33%) for the IgH/MYC probe. The range of single fusion signals was 30–44% (mean 38%, median 38%) for the IgH/bcl-2 probe, 34–51% (mean 47%, median 50%) for the IgH/CCND1 probe and 32–64% (mean 44%, median 49%) for the IgH/MYC probe.
Representative examples of the fluorescent tissue appearance and histogram analyses are shown for a normal lymph node compared to mantle cell lymphoma and for normal lymph node compared to Burkitt lymphoma (Composite Figures 1 and 2, respectively). As seen in the histograms, a normal lymphoid tissue specimen shows a peak at ‘0’ for fusions (Figures 1a and 2a, right lower panel). In contrast, positive cases show a characteristic ‘shift to the right’ with peaks at ‘1 and 2’ for the increased number of fusion signals (Figures 1b and 2b, right lower panel).
Diagnostic Sensitivity
Automated tissue fluorescence in situ hybridization analysis identified the presence of the t(11;14) in 100% of the tested mantle cell lymphoma cases with interpretable signals (7/7), the t(8;14) in 100% of the tested Burkitt lymphomas with interpretable signals (9/9), and the t(14;18) in 91% of the tested follicular lymphomas with interpretable signals (10/11) (Table 3). The cases were identified as positive for the translocation by detecting an increased number of fusion signals compared to normal controls, using dual fusion detection strategy fluorescence in situ hybridization probes (as described above in automated analysis). Conversely, none of the twenty non-neoplastic lymph nodes showed evidence of increased fusion signals with the three probe sets. Two cases each of follicular lymphoma and Burkitt lymphoma showed no interpretable hybridization signals. Although the overall test sample size is small, we verify and validate (with normal control specimens) that this automated technique is accurate and sensitive in its detection of diagnostically and potentially prognostically relevant fusion products.
Discussion
Our study demonstrates that automated analysis of fluorescence in situ hybridization signals on fixed, whole, paraffin-embedded, tissue sections is available, accurate, sensitive and alleviates the difficulties associated with scoring of truncated nuclei in tissue sections. This technique not only plays a critical role in the diagnosis and prognosis of B-cell lymphomas, as shown here, but is potentially applicable to a wide variety of hematolymphoid and non-hematolymphoid disorders. Furthermore, this plays a critical role in the evaluation of ‘real-time’ clinical cases when there is insufficient fresh or unfixed material for conventional cytogenetics and/or fluorescence in situ hybridization. Also, a variety of tissue fixatives (formalin, acetic acid zinc formalin, B5, decalcified bone marrow core biopsies) can be utilized. The advantages and disadvantages of fluorescence in situ hybridization on fixed whole tissue sections are summarized in Table 4.
Our initial studies show that this technique of automated analysis of tissue fluorescence in situ hybridization can positively identify all tested cases of mantle cell lymphoma with the CCND1-IgH fusion (100% sensitivity), all cases of Burkitt lymphoma with MYC-IgH fusion (100% sensitivity) and detect the IgH-bcl-2 fusion in 91% of follicular lymphomas (Table 2). Although our sample size for each type of lymphoma is small, we are able to definitively validate that automated analysis detects fusions in all cases where a fusion is present. In follicular lymphoma, it is well known in the literature that not all cases demonstrate the t(14;18). Therefore, it is not unexpected that our detection rate is below 100%. There was 100% cytogenetic concordance between the automated analysis and those seen at diagnosis (all cases of mantle cell lymphoma and Burkitt lymphoma, cytogenetic data was available for only two cases of follicular lymphoma). Also, although not presented in this study, it is possible (using the same classifier as for fusion counts) to detect abnormalities of ploidy that may be present. For example, identification of three copies of the MYC gene with no fusions (using the t(8;14) probe set), suggests the possibility of an alternate translocation partner in Burkitt lymphoma.
We also demonstrate variable success with fluorescence in situ hybridization hybridization on several types of tissue fixatives. The majority of our tested tissue sections were formalin or acetic acid zinc formalin-fixed with hybridization rates approaching 100% (Table 2). Importantly, fluorescence in situ hybridization also appears to work about half the time on B5-fixed tissues or decalcified bone marrow core biopsy specimens, although only a handful of cases were evaluated (4 and 7, respectively). This certainly presents the option for fluorescence in situ hybridization testing in cases when no other, more-desirable (eg fresh cells, formalin fixed or clot section) specimen, is available.
Fluorescence in situ hybridization on whole tissue sections is powerful in that it allows the pathologist/clinician to gain valuable diagnostic and prognostic genetic information from a fixed specimen. Such information can be gained by performing fluorescence in situ hybridization on either isolated interphase nuclei6 or intact tissue sections.8, 9, 10, 11 In the technique described by Paternoster and colleagues,6 individual whole nuclei are extracted from the particular areas of interest in the block, and then subjected to typical manual fluorescence in situ hybridization interphase analysis. In our technique, a whole tissue section is utilized which preserves the architecture and distribution of the areas of interest and eliminates the additional step of nuclei extraction. Although we did not do a specific comparison of manual vs automated analysis, it is clear that there are difficulties that are regularly anticipated with the manual process. Just on low power scanning for interpretable areas, these difficulties include areas with too dense of nuclei (thick section, small tumor cells, etc.) to accurately ascertain individual signals per nucleus, overlapping nuclei (as a result of tissue architecture) and truncated nuclei (as a result of the process of tissue sectioning itself). These troublesome features, inherent to performing fluorescence in situ hybridization on sectioned tissues, are addressed by the MetaSystems™ automated image capture and analysis system by enumerating the signals seen per area or ‘tile’, over many areas of tumor tissue. The tile size is set manually by the interpreter and reflects the average size of the tumor cells in the particular case.
Fluorescence in situ hybridization on fixed tissue specimens is an important tool in the clinical evaluation of hematolymphoid disorders, but also applies to a variety of surgical pathology entities. This technique may be used in routine clinical diagnosis, but also serves as a valuable research tool for analysis of archival tissue specimens. New insights into underlying genetic abnormalities may be investigated utilizing materials from previously diagnosed cases. Such broad applicability will undoubtedly influence our understanding of genetics in disease and will contribute to our progress in realizing ‘molecularly-defined’ entities.
References
Jaffe ES, Harris NL, Stein H, et al. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon, 2001.
Bagg A, Braziel RM, Arber DA, et al. Immunoglobulin heavy chain gene analysis in lymphomas: a multi-center study demonstrating the heterogeneity of performance of polymerase chain reaction assays. J Mol Diagn 2002;4:81–89.
Rimokh R, Berger F, Delsol G, et al. Detection of the chromosomal translocation t(11;14) by polymerase chain reaction in mantle cell lymphomas. Blood 1994;83:1871–1875.
Raynaud SD, Bekri S, Leroux D, et al. Expanded range of 11q23 breakpoints with differing patterns of cyclin D1 expression in B-cell malignancies. Genes Chromosom Cancer 1993;8:80–87.
Jian-Yong L, Fanny G, Anne M, et al. Detection of Translocation t(11;14)(q13;q32) in Mantle Cell Lymphoma by Fluorescence in Situ Hybridization. Am J Pathol 1999;154:1449–1452.
Paternoster SF, Brockman SR, McCLure RF, et al. A new method to extract nuclei from paraffin-embedded tissue to study lymphomas using interphase fluorescence in situ hybridization. Am J Pathol 2002;160:1967–1972.
Matsumoto Y, Nomura K, Matsumoto S, et al. Detection of t(14;18) in follicular lymphoma by dual-color fluorescence in situ hybridization on paraffin-embedded tissue sections. Cancer Genet Cytogenet 2004;150:22–26.
Nomura K, Sekoguchi S, Ueda K, et al. Differentiation of follicular from mucosa-associated lymphoid tissue lymphoma be detection of t(14;18) on single-cell preparations and paraffin-embedded sections. Genes Chromosomes Cancer 2002;33:213–216.
Nomura K, Yoshino T, Nakamura S, et al. Detection of t(11;18)(q21;q21) in marginal zone lymphoma of mucosa-associated lymphoid tissue type on paraffin-embedded tissue sections by using fluorescence in situ hybridization. Cancer Genet Cytogenet 2003;140:49–54.
Hirose Y, Masaki Y, Karasawa H, et al. Incidence of diffuse large B-cell lymphoma of germinal center B-cell origin in whole diffuse large B-cell lymphoma: tissue fluorescence in situ hybridization using t(14;18) compared with immunohistochemistry. Int J Hematol 2005;81:48–57.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reichard, K., Hall, B., Corn, A. et al. Automated analysis of fluorescence in situ hybridization on fixed, paraffin-embedded whole tissue sections in B-cell lymphoma. Mod Pathol 19, 1027–1033 (2006). https://doi.org/10.1038/modpathol.3800630
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800630
