
Abstract
p53 mutations have been implicated in the development of esophageal malignancies. The purpose of this study was to assess more accurately the incidence and types of p53 mutations in Barrett's esophagus (BE) with and without dysplasia and in esophageal adenocarcinoma, using pure preparations of epithelial cells obtained by laser capture microdissection (LCM). Assays were performed on paraffin-embedded tissue samples of normal antrum and premalignant and malignant esophageal samples from 57 patients, including 16 controls, 10 with BE metaplasia alone, 20 with BE-associated dysplasia, and 11 with BE-associated adenocarcinoma. All tissues were processed for LCM. DNA was extracted from isolated cells, and polymerase chain reaction (PCR) was performed using oligonucleutide primers for exons 5–8 of p53. PCR products were processed for DNA sequencing. p53 sequence abnormalities were identified in 2/16 cases of normal antrum and regenerative/chemical gastritis, 1/10 cases of BE, 1/20 cases of BE with dysplasia, and 2/11 cases of adenocarcinomas. The abnormalities occurred in exons 7 and 8 in the form of point mutations. Our results, using LCM, show that p53 gene mutations are relatively rare in esophageal preneoplastic and neoplastic conditions. Only point mutations were detected, but no deletions/insertions were identified.
Similar content being viewed by others

Main
Barrett's esophagus (BE) develops through a metaplastic process in which an abnormal columnar epithelium replaces the normal stratified squamous epithelium of the distal esophagus.1, 2, 3 Furthermore, this lesion predisposes to the development of dysplasia and adenocarcinoma of the esophagus.
The prediction of which patients with Barrett's metaplasia will progress to malignancy is difficult. A better genetic characterization of this condition may help clinicians to determine the risk of cancer development and to elaborate an adequate preventive strategy as well as better treatment and follow-up plans.
Mutations of the tumor-suppressor gene p53 have been implicated in the pathogenesis of esophageal carcinomas. The purpose of this study was to assess accurately the incidence and types of p53 mutations in BE with and without dysplasia and in esophageal adenocarcinoma, using pure preparations of epithelial cells obtained by laser capture microdissection (LCM).
Materials and methods
Tissue Samples
A total of 57 formalin-fixed paraffin-embedded samples were retrieved from the surgical pathology files of Beth Israel Deaconess Medical Center, Boston, MA, USA, with patients' ages ranging from 33 to 84 years. Specimens were obtained from patients with normal antrum (10), regenerative/chemical gastritis (six), BE without dysplasia (10), BE with dysplasia (20), and adenocarcinoma of esophagus (11).
Histopathologically, regenerative/chemical gastritis is associated with regeneration and hyperplasia of the surface and foveolar mucous cells as well as the absence of neutrophils in the lamina propria.4 Dysplastic specimens are classified as ‘low-grade dysplasia’ (with decreased mucus secretion, crowding of slender columnar cells with pseudostratified nuclei, and occasional mitosis; pleomorphism is absent or mild; the glands retain the normal contour but may be enlarged), and ‘high-grade dysplasia’ (where the criteria include moderate pleomorphism, plump cells, marked reduction of mucus secretion, and frequent mitosis; the glands show budding, branching, crowding, and intraluminal infolding). Adenocarcinomas are graded as well and poorly differentiated.
Samples were used for LCM, polymerase chain reaction (PCR), and DNA sequencing (Figure 1).

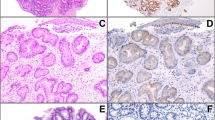
Hematoxylin and eosin-stained tissue sections of: (a) normal antrum; (b) regenerative/chemical gastritis; (c) Barrett's esophagus; (d) dysplasia; (e) adenocarcinoma of esophagus (× 10).
LCM
All 57 formalin-fixed paraffin-embedded samples were recut on glass slides coated with LPC-membrane (PEN Foil 1.35 μm, P.A.L.M. Microlaser Technologies, Bernried, Germany) and deparaffinized. Slides were then stained with toluidine blue and used for LCM (Figure 2). Pure cell populations of normal antrum, regenerative gastric glands, and Barrett metaplastic, dysplastic, and adenocarcinomatous glands were obtained from our samples by LCM (P.A.L.M., Microlaser Technologies).

Toluidine-blue-stained sections of normal antrum before (a) and after (b) LCM.
DNA Extraction
DNA was extracted from isolated cells using the PURGENE DNA Isolation Kit protocol and resuspended in 50 μl of ddH2O (Gentra System, Minneapolis, MN, USA).
PCR Amplification
Exons 5–8 of the p53 gene (the sites of most somatic p53 mutations) were amplified by PCR using amplification primers from published sequences5 as follows: exon 5: 5′-ttcctcttcctgcagtactc-3′ and 5′-cagctgctcaccatcgct-3′, exon 6: 5′-cactgattgctcttaggt-3′ and 5′-agttgcaaaccagacctc-3′, exon 7: 5′-gttggctctgactgtaccaccat-3′ and 5′-gctcctgacctggagtct-3′, and exon 8: 5′-cctatcctgagtagtggt-3′ and 5′-tcctgcttgcttacctcgct-3′. The size of PCR DNA fragments were: 190, 126, 96, and 165 bp, respectively.
An amount of 10 μl of DNA was subjected to 40 cycles of PCR in a volume of 50 μl containing 50 ng of each oligonucleotide primer, 0.5 UTaq DNA polymerase, 8 μl of dNTP at 1.5 mM, 24.5 μl dH2O, and 5 μl of 10 × PCR buffer containing 1.5 mM MgCl2 (Applied Biosystems, Foster City, CA, USA). PCR conditions were 94°C for 1 min, 53°C for 2 min, and 72°C for 1 min.
DNA fragments from PCR were then purified using the QIAquick PCR Purification Kit Protocol (QIAGEN, Valencia, CA, USA), and electrophoresed on 2% agarose gel.
DNA Sequence Analysis
Amplified DNA fragments were sequenced using an ABI prism 373 Sequencer (Applied Biosystems) in both directions.
Statistical Analysis
A two-way contingency table with Fisher's exact correction (StatView for Windows, SAS Institute Inc., version 5.0.1, Cary, NC, USA) was used to compare the incidence of p53 mutations among the lesions studied.
Results
Point mutations of p53 were identified in six of the 57 cases studied (Table 1). They were detected in two of the 16 (12.5%) normal/regenerative stomach; in one of the 10 (10%) BE; in one of the 20 (5%) low-grade and high-grade dysplasia; and in two of the 11 (18%) esophageal adenocarcinoma. There was no significant difference in the incidence of point mutations between normal/regenerative antrum and BE without dysplasia (P>0.05), between BE without dysplasia and BE with dysplasia (P>0.05), and between BE with dysplasia and adenocarcinoma (P>0.05). These results suggest that p53 gene mutations are relatively rare in esophageal preneoplastic and neoplastic conditions. In all these cases, p53 gene mutations were in the form of point mutations involving exons 7 and 8 (Table 2, Figure 3). Deletions/insertions were not identified.

DNA sequence of exon 8 of p53 gene: point mutation in codon 300 in a case of well-differentiated adenocarcinoma (case #54) with C → T substitution (a), compared to normal sequence (b).
Of the two mutations present in normal antrum, one of them was a ‘silent’ mutation and did not result in a change of amino-acid sequence (Table 2, case 14). All other mutations resulted in a change of amino-acid sequence, one in normal antrum, one in BE, one in dysplastic Barrett's epithelium, and two in invasive adenocarcinomas (Table 2).
Discussion
Carcinogenesis in metaplastic esophageal columnar cells begins with genetic alterations that activate proto-oncogenes, disable tumor suppressor genes, or involve both mechanisms.6 The evolution of genetic changes leading from BE to adenocarcinoma is incompletely understood. However, several studies have shown that this neoplastic progression includes alterations in the tumor suppressor genes p53 and p16 and nonrandom losses of heterozygosity (LOH).7, 8, 9, 10 In addition to these changes, tetraploid populations are observed in more than 90% of adenocarcinomas.8
A relatively low frequency of p16 (retinoblastoma) gene mutation has been reported in patients with BE and esophageal adenocarcinoma.11 Cytogenetic alterations have also been studied by comparative genomic hybridization (CGH). Frequent losses on the Y chromosome of 4q, 5q, 9p, 18q, 7q, and 14q and gains on 8q, 20q, 2p, 7p, 10q, 6p, 15q, and 17p were detected in metaplastic BE and esophageal adenocarcinoma, and a correlation between an increase in abnormalities and progression toward dysplasia and adenocarcinoma has been noted.12 DNA content, measured by flow cytometry, has been also the subject of numerous studies, reporting that aneuploidy and increased G2M/tetraploid populations may increase along with increasing dysplasia, but the results have been conflicting.13, 14
The prevalence of p53 gene mutation in esophageal carcinoma reported in the literature ranges from 42 to 67%,6, 15, 16, 17 with the exception of two reports in which a low prevalence (8%) (18) and a higher prevalence (84%),19 respectively, were observed. The prevalence of protein accumulation is more variable, ranging from 34 to 87%.15, 16, 17 The discordance between immunohistochemistry (high frequency of positively staining cases) and molecular techniques (relatively low number of p53 gene mutations) reported in the literature ranged between 24 and 45% of cases16, 20, 21, 22 of esophageal carcinoma. This discordance between p53 phenotype/genotype expression raises a problem in the interpretation of p53 accumulation demonstrated by immunohistochemistry as an indirect sign of p53 mutations.
In our study, using LCM, we provided a more precise assessment of the incidence and types of p53 mutations in BE with and without dysplasia and in esophageal adenocarcinoma. Indeed, selection of areas of interest and precise microdissection of those focal areas permitted us to obtain pure epithelial cell preparations. Our results, contrary to the majority of publications, did not show a significant increase in the incidence of p53 gene mutation in premalignant and malignant esophageal lesions in the exons that we screened. This is probably due to the purity of our cell population obtained by selection of specific cells by LCM, and direct sequencing of PCR products in both directions, rather than using indirect techniques.7, 12, 15, 16, 18, 23, 24, 25, 26, 27, 28, 29 Therefore, the role of p53 mutation as a prognostic factor in progression of metaplastic BE toward esophageal adenocarcinoma is not certain. Since the molecular basis of p53 function and mutations is not fully understood, a better evaluation of the biological properties of different p53 mutations is needed in order to interpret the results. Also, the hypothesis of mutations arising at different stages in the evolution of the BE is of concern, since we found no statistically significant increase in mutations compared to controls. However, our findings are based on a relatively limited number of cases, and a large multicenter study is needed to better evaluate the role of p53 alterations in progression of BE and as a prognostic factor.
In conclusion, the results of our study show that mutations of p53 using exons 5–8 are relatively rare in esophageal preneoplastic and neoplastic conditions and therefore are of limited use as a marker to study disease progression.
References
Phillips RW, Wong RKH . Barrett's esophagus. Natural history, incidence, etiology and complications. Gastroenterol Clin North Am 1991;20:791–816.
Spechler SJ, Zeroogian JM, Antonioli DA, et al. Prevalence of metaplasia at the gastro-oesophageal junction. Lancet 1994;344:1533–1536.
Spechler SJ . Laser photoablation of Barrett's epithelium: burning issues about burning tissues. Gastroenterology 1993;104:1855–1858.
Ming S-C . Adenocarcinoma and other epithelial tumors of the esophagus. In: Ming S-C, Goldman H (eds). Pathology of the Gastrointestinal Tract, 2nd edn. Williams&Wilkins: Baltimore, MD, 1998, pp 503–509.
Gramlich TL, Fritsch C, Cohen C, et al. Oncogene expression and amplification in Barrett adenocarcinoma. Int J Surg Pathol 1997;4:203–212.
Souza RF, Meltzer SJ . The molecular basis for carcinogenesis metaplastic columnar-lined esophagus. Gastroenterol Clin North Am 1997;26:583–597.
Neshat K, Sanchez CA, Galipeau PC, et al. p53 mutations in Barrett's adenocarcinoma and high-grade dysplasia. Gastroenterology 1994;106:1589–1595.
Galipeau PC, Cowan DS, Sanchez CA, et al. 17p (p53) allelic losses, 4N (G2/tetraploid) populations, and progression to aneuploidy in Barrett's esophagus. Proc Natl Acad Sci USA 1996;93:7081–7084.
Wong DJ, Barrett MT, Stoger R, et al. p16NK4a promoter is hypermethylated at a high frequency in esophageal adenocarcinomas. Cancer Res 1997;57:2619–2622.
Galipeau PC, Prevo LJ, Sanchez CA, et al. Clonal expansion and loss of heterozygosity at chromosomes 9p and 17p in premalignant esophageal (Barrett's) tissue. J Natl Cancer Inst 1999;91:2087–2095.
Krishnadath KK, Reid BJ, Wang KK . Biomarkers in Barrett esophagus. Mayo Clin Proc 2001;76:438–446.
Walch AK, Zitzelsberger HF, Bruch J, et al. Chromosomal imbalances in Barrett's adenocarcinoma and the metaplasia–dysplasia–carcinoma sequence. Am J Pathol 2000;156:555–566.
Reid BJ, Haggitt RC, Rubin CE, et al. Flow cytometry complements histology in detecting patients at risk for Barrett's adenocarcinoma. Gastroenterology 1987;93:1–11.
Fennerty MB, Sampliner RE, Way D, et al. Discordance between flow cytometric abnormalities and dysplasia in Barrett's esophagus. Gastroenterology 1989;97:815–820.
Bian YS, Osterheld MC, Bosman FT, et al. p53 gene mutation and protein accumulation during neoplastic progression in Barrett's esophagus. Mod Pathol 2001;14:397–403.
Coggi G, Bosari S, Roncalli M, et al. p53 protein accumulation and p53 gene mutation in esophageal carcinoma. Cancer 1997;79:425–432.
Kubba AK, Pool NA, Watson A . Role of p53 assessment in management of Barrett's esophagus. Dig Dis Sci 1999;44:659–667.
Casson AG, Mukhopadhyay T, Cleary KR, et al. p53 gene mutations in Barrett's epithelium and esophageal cancer. Cancer Res 1991;51(16): 4495–4499.
Audrezet MP, Robaszkiewicz M, Mercier B, et al. TP53 gene mutation profile in esophageal squamous cell carcinomas. Cancer Res 1993;53:5745–5749.
Gao H, Wang L-D, Zhou Q, et al. p53 tumor suppressor gene mutation in early esophageal precancerous lesion and carcinoma among high-risk populations in Henan, China. Cancer Res 1994;54:4342–4346.
Wagata T, Shibagaki I, Imamura M, et al. Loss of 17p, mutations of the p53 gene, and overexpression of p53 protein in esophageal squamous cell carcinomas. Cancer Res 1993;53:846–850.
Moore JH, Lesser EJ, Erdody DH, et al. Intestinal differentiation and p53 gene alterations in Barrett's esophagus and esophageal adenocarcinoma. Int J Cancer 1994;56:487–493.
Casson AG, Manolopoulos B, Troster M, et al. Clinical implications of p53 gene mutation in the progression of Barrett's epithelium to invasive esophageal cancer. Am J Surg 1994;167:52–57.
Prevo LJ, Sanchez CA, Galipeau PC, et al. p53-mutant clones and field effects in Barrett's esophagus. Cancer Res 1999;59:4784–4787.
Schneider PM, Casson AG, Levin B, et al. Mutations of p53 in Barrett's esophagus and Barrett's cancer: a prospective study of ninety-eight cases. J Thorac Cardiovasc Surg 1996;111:323–333.
Dunn JR, Garde J, Dolan K, et al. The evolution of loss of heterozygosity on chromosome 17 during the progression to Barrett's adenocarcinoma involves a unique combination of target sites in individual specimens. Clin Cancer Res 2000;6:4033–4042.
Bhargava P, Eisen GM, Holterman DA, et al. Endoscopic mapping and surrogate markers for better surveillance in Barrett esophagus. Am J Clin Pathol 2000;114:552–563.
Reid BJ, Prevo LJ, Galipeau PC, et al. Predictors of progression in Barrett's esophagus II: baseline 17p (p53) loss of heterozygosity identifies a patient subset at increased risk for neoplastic progression. Am J Gastroenterol 2001;96:2839–2848.
Giménez A, Minguela A, Parrilla P, et al. Flow cytometric DNA analysis and p53 protein expression show a good correlation with histologic findings in patients with Barrett's esophagus. Cancer 1998;83:641–651.
Author information
Authors and Affiliations
Corresponding author
Additional information
The study was presented at the United States and Canadian Academy of Pathology meeting in Washington, DC, March 2003.
Rights and permissions
About this article
Cite this article
Djalilvand, A., Pal, R., Goldman, H. et al. Evaluation of p53 mutations in premalignant esophageal lesions and esophageal adenocarcinoma using laser capture microdissection. Mod Pathol 17, 1323–1327 (2004). https://doi.org/10.1038/modpathol.3800231
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800231
Keywords
This article is cited by
-
Targeted genetic and epigenetic profiling of esophageal adenocarcinomas and non-dysplastic Barrett’s esophagus
Clinical Epigenetics (2022)
