
Abstract
Epstein–Barr (EBV) virus is associated with malignancies such as lymphoma and carcinoma. Infection of cells with EBV may result in either lytic infection with production of viral particles, characterized by the presence of linear DNA forms, or latent infection, characterized by either episomal or integrated DNA forms. To examine whether the different lytic and latent EBV DNA forms can reliably be distinguished in single human cells, in situ hybridization was performed in EBV-positive cell lines. Immunocytochemistry and Southern blot analysis were performed supplementary to in situ hybridization. In latent infection, three in situ hybridization patterns were observed: large-disperse (episomal), small-punctate (integrated) and combined (both), signal types 1, 2 and 3 respectively. These were associated with expression of latent membrane protein 1, but not with Z fragment of Epstein–Barr replication activator or viral capsid antigen. In lytic infection, three additional in situ hybridization patterns were observed: nuclear membrane associated, bubble (filling up the nucleus) and spillover (covering the lysed cells) signals types 4, 5 and 6 respectively. Signal types 4 and 5 were associated with expression of latent membrane protein 1 and Z fragment of Epstein–Barr replication activator but not viral capsid antigen, whereas type 6 was associated with expression of viral capsid antigen only. Southern blot analysis confirmed these results; however, low copy numbers of integrated virus were often missed by Southern blot, confirming that in situ hybridization is more sensitive in determining the presence of all types of EBV DNA. In situ hybridization may prove useful in rapidly screening large series of tissue microarrays and other clinical specimens for the presence of lytic or latent EBV.
Similar content being viewed by others

Main
Epstein–Barr Virus (EBV) has been implicated as a cause of malignant transformation in a number of lymphoid and nonlymphoid cell types. The first association of EBV with cancer was made in 1964 and coincided with discovery of the virus by Epstein, Achong and Barr in electron micrographs of cells cultured from patients with endemic Burkitt's lymphoma.1, 2 EBV is associated with over 90% of cases of endemic Burkitt's lymphoma, 30–50% of Hodgkin's lymphomas, and up to 50% of non-Hodgkin's lymphoma in immunosuppressed patients.3 EBV carries a set of latent genes that, when expressed in resting B cells, induces cell proliferation and thereby increase the chance of successful viral colonization of the B-cell system and the establishment of persistence during primary infection. However, if this cell proliferation is not controlled by the immune system, or if it is accompanied by additional genetic events within the infected cell, it can lead to malignancy.4, 5
In the latest International Agency for Research on Cancer monograph of the World Health Organization EBV was classified as a group 1 carcinogen, an indication that there is the strongest possible evidence linking it to human cancer,6 and making recognition of the presence of EBV DNA within cells important. Infection of cells with EBV may result in production of lytic virus, which is characterized by the presence of linear viral DNA. Alternatively, it may result in viral latency, characterized by persistence of the viral genome in episomal form or integrated into the host cell genome. With viral replication, progeny virions are released, resulting in characteristic cytopathic effects such as ballooning of the cell, fragmentation of nuclear DNA and cell lysis.7 Viral persistence in a cell may result in cellular transformation, providing the basis for the establishment of in vitro or in vivo immortalized lymphoblastoid cell lines.8, 9, 10, 11
The majority of latently infected EBV-positive cell lines contain viral genomes as independent episomal structures; however, evidence for integration of the EBV genome into human chromosomes has been demonstrated in the cell lines Namalwa, IB4, EB2 and several lymphoblastoid cell lines.12, 13, 14, 15, 16, 17, 18 During latent infection, EBV can be reactivated and lytic infection initiated. Although spontaneous production of virus is more frequent in so-called producer cell lines such as B95-8, a small proportion of cells (<10%) produce and release virus in latently infected transformed lymphoblastoid cell lines.19, 20, 21
Latently infected cells express a particular set of viral genes, which encode characteristic nuclear and integral membrane proteins such as Epstein–Barr nuclear antigens and latent membrane proteins. The BZLF1 gene product Z fragment Epstein–Barr-replication activator initiates the lytic cycle, while late viral replication is characterized by expression of viral capsid antigen.22, 23
In order to investigate whether different forms of intracellular EBV DNA can be recognized and discriminated using in situ hybridization, cell lines in which the virus persists in latent, lytic, or both states were analyzed. These EBV positive cell lines contain well-defined numbers of EBV genomes and both the mode of infection and physical state of viral DNA is well characterized. We confirmed the state of EBV infection recognized by in situ hybridization using two complementary approaches; Southern blot analysis of viral genomes and determination of viral protein expression by immunocytochemistry.
Materials and methods
Cell Lines and DNA probes
The cell lines EB2, Daudi, Namalwa, Raji and the producer cell line B95-8 were obtained from the American Type Culture Collection (ATCC). The three lymphoblastoid cell line (LCL-112, LCL-36, and LCL-MD) used in this study have been characterized elsewhere by cytogenetic analysis.4 Cell cultures, chromosome preparations and cytospins were made according to standard procedures. The biotinylated EBV-BamHIW fragment (ENZO Biochemical, Inc., New York, NY, USA) and the XhoIa and EcoRI1 terminal fragments (kindly provided by Dr N Raab-Traub) were used for in situ hybridization and Southern blot analysis, respectively.
Immunocytochemistry
Immunocytochemistry was performed either alone or prior to fluorescence in situ hybridization. Cytospin preparations were fixed for 10 min in acetone, briefly washed in phosphate-buffered saline (PBS) and blocked for 10 min in PBS containing 0.1% bovine serum albumin (BSA/PBS). Cells were incubated with antibodies directed against Z fragment of Epstein–Barr replication activator (1:20, DAKO, Denmark), latent membrane protein 1 (1:25, DAKO) or viral capsid antigen (1:100, Virotech, USA) and with rabbit anti-mouse-TRITC (1:100, DAKO). Slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and mounted in the Vectashield mounting medium (Vector laboratories Inc., Burlingame, CA, USA). For double immunoenzymatic staining, the first antibody was detected with rabbit anti-mouse-peroxidase (1:100, DAKO) followed by development with 3.3% diaminobenzidine (DAB) (Sigma, St Louis, MO, USA). The second antibody was detected by the alkaline phosphatase–anti-alkaline phosphatase (APAAP) method. Alkaline-phosphatase was developed with fast red TR salt (Sigma). Slides were counterstained in Mayer's hemalum (Merck, Germany) and mounted with the Glycergel mounting medium (DAKO). All antibodies were diluted in 3% BSA/PBS and all slides incubated at 37°C in a humidified chamber for 1 h. Slides were washed twice (10 min each) in 0.1% BSA/PBS between each incubation step.
Fluorescence In Situ Hybridization and Peroxidase-Based In Situ Hybridization
Slides were treated with DNase-free RNase (100 μg/ml) for 1 h at 37°C to avoid DNA–RNA crosshybridizations. Subsequently, the slides were fixed with 4% paraformaldehyde/1% methanol in PBS for 30 min at 4°C prior to fluorescence in situ hybridization. An amount of 10 μl hybridization mixture containing 50% deionized formamide (Merck), 10% dextran sulfate (Pharmacia AB, Sweden) in 2 × SSC, 0.3 μg/μl sonicated salmon sperm DNA (Sigma) and 0.2 ng/μl biotinylated EBV-BamHIW fragment (ENZO, USA) was applied to each slide for EBV fluorescence in situ hybridization. Denaturation, hybridization, posthybridization washing steps, immunocytochemical detection, counterstaining, and fluorescence microscopy was performed as described elsewhere.3, 14 Peroxidase-based in situ hybridization was performed in a manner similar to that of fluorescence in situ hybridization, with the exception that immunocytochemical detection of the biotinylated probe was carried out using 1:20 mouse anti-biotin, 1:100 rabbit anti-mouse peroxidase and 1:100 swine anti-rabbit peroxidase (all DAKO). Peroxidase activity was detected with diaminobenzidine (DAB, Sigma) and 3% hydrogen peroxide (H2O2). Slides were counterstained in Mayer's hemalum (Merck), dehydrated and mounted with entellan (Merck). Hybridization signals were evaluated by conventional light microscopy. For combined assessment, immunocytochemistry was performed on the first day of analysis, followed by fluorescence in situ hybridization, completed on the second day.
Southern Blot Analysis
High molecular weight DNA was extracted using the standard high-salt extraction method. For each cell line, 10 μg of DNA was digested to completion with BamHI and subjected to electrophoresis through a 0.8% agarose gel. Southern blot analysis was performed according to standard procedures. For filter hybridization, probes flanking the left (EcoRI1) and the right (XhoIa) terminal repeats of the viral genome were labeled with 32P dCTP by the random primer method. Autoradiography was carried out at −70°C for 5 days.
Criteria for Discriminating Latent and Lytic Intracellular EBV DNA Forms
For Southern blot analysis, adapted interpretation criteria were used.24 The signal types and physical state of intracellular latent EBV DNA determined by in situ hybridization analysis were assessed according to proposals for human papilloma virus.25, 26, 27, 28 EBV copy number and state of viral infection of the analyzed cell lines were evaluated in accordance with criteria published in detail elsewhere, for example the number of viral genomes present were counted directly using light microscopy.14, 29, 30, 31, 32
Results
Sensitivity of Fluorescence and Peroxidase-Based In Situ Hybridization
Experiments were carried out to determine the sensitivity of fluorescence and peroxidase-based in situ hybridization. No difference in quality and in evaluation of EBV DNA signals was observed using either in situ hybridization methodology. Fluorescence in situ hybridization, however, proved to be the method of choice when combined with immunocytochemistry. EBV DNA signals were recognized in cells with numbers ranging from two (Namalwa) to over 100 (Daudi, B95-8) copies per cell, although in the latter case reliable estimation of the number of viral genomes was difficult. All results are summarized in Table 1.
Characterization of Latent EBV Infection (Signal Types 1–3)
Large, disperse intranuclear hybridization signals were observed, which we termed large-disperse or type 1 signal (Figure 1a–d). These occurred in the human cell lines EB2, Raji, Daudi and the lymphoblastoid cell lines LCL-36, LCL-MD and LCL-112, all of which are documented to contain episomal EBV DNA. The number and appearance of these hybridization signals varied considerably between cell lines and between individual cells within a particular cell line. The intranuclear signal type termed small punctate or type 2 had sharp contours, and frequently occurred in doublets reflecting signals on both sister chromatids at the chromosomal position 1p35 (Figure 1b, e). This signal type is a typical feature of the cell line Namalwa that is documented to contain two integrated viral genomes, and showed pairs of closely adjacent spots in 85% of interphase nuclei, suggesting integration into each of the pair of sister chromatids. About 10% of nuclei displayed only a single spot, which may have reflected close juxtaposition of signals. The remaining 5% of cells showed two pairs of intranuclear spots, suggestive of replication of cellular (and integrated viral) DNA such as occurs in the G2 phase of the cell cycle. Type 1 signal was never found in the cell line Namalwa, which is documented to completely lack episomal forms. With the exception of Namalwa, a mixture of in situ hybridization signals was observed in all cell lines known to exhibit latent infection, and we termed this combined or type 3 signal (Figure 1c, f, g). In addition to large-disperse in situ hybridization signals, representing episomal virus, small punctate signals were observed. Cells in metaphase had episomes in multiple copies, which appeared as multiple randomly distributed hybridization signals. Signals on both sister chromatids were present at the same chromosome band with an average of seven to about 30 integrated copies per cell, depending on the cell line, consistent with the presence of integrated viral DNA. These results are summarized in Table 1.

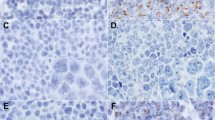
Latent virus: Large-disperse signal type 1: episomal viral DNA seen as large numbers of signals clustered together in a Daudi interphase nucleus (a) and a Daudi metaphase cell (d) (fluorescence in situ hybridization). Small-punctate signal type 2: one or two (doublets) sharp signals as seen in Namalwa interphase nuclei (peroxidase-based in situ hybridization) (b) and a Namalwa metaphase cell with integration of two EBV genomes at chromosomal band 1p35 (fluorescence in situ hybridization, yellow dots, e). Combined signal type 3: Coexistence of both signal types 1 and 2 as seen in Daudi interphase nuclei (c) and Daudi metaphase chromosomes in which arrows indicate some integrated copies (f) (peroxidase-based in situ hybridization). Raji interphase nuclei demonstrating some integrated and high numbers of episomal copies (fluorescence in situ hybridization) (g). LCL-112 (h) and EB2 (i) interphase nuclei demonstrating type 3 viral signals (green) and latent membrane protein 1 expression (red) as seen by fluorescence in situ hybridization and immunocytochemistry. Lytic Virus: Nuclear membrane associated signal type 4: viral DNA is localized near the nuclear periphery in LCL-112 interphase nuclei (j) and an EB2 interphase nucleus (m) with type 4 viral signals (green) and latent membrane protein 1 expression (red) as seen by fluorescence in situ hybridization and immunocytochemistry. Bubble signal type 5: an LCL-112 cell by using peroxidase-based in situ hybridization (brown signals, k) or fluorescence in situ hybridization (green signals, n) demonstrating round three-dimensional formations representing cells filled with virus on the verge of egress. A B95-8 cell (p) demonstrating coexpression of latent membrane protein 1 (brown) and Z fragment of Epstein–Barr replication activator (red). Spillover signal type 6: A B95-8 cell lysed and completely covered by virus demonstrated by pox-ISH (l) or by fluorescence in situ hybridization and immunocytochemistry where viral DNA is green and Z fragment of Epstein–Barr replication activator red (o), or viral DNA is green and viral capsid antigen red (r). An LCL-112 cell (q) demonstrating coexpression of Z fragment of Epstein–Barr replication activator (red) and viral capsid antigen (brown).
Characterization of Lytic EBV Infection (Signal Types 4–6)
In the cell lines B95-8, Daudi, EB2 and LCL-112, three additional hybridization patterns were identified. The first lytic signal type was located at the nuclear periphery in compact, diffuse round formations, indicating the presence of large numbers of EBV DNA copies; this was termed nuclear membrane associated or type 4 signal (Figure 1j). In the second type, individual nuclei were completely covered by signal in a three-dimensional manner, indicating the presence of vast quantities of EBV DNA; we termed this pattern bubble or type 5 signal (Figure 1k, n). Finally, viral signals were spread in massive numbers within and around disrupted cells, which we termed spillover or type 6 signal (Figure 1l). Previous characterization of cell lines suggests that these signal types correspond to the switch from latent to lytic infection, production of viral DNA, and release of mature virions, respectively. The three signal types representing lytic infection were present in >10% of cells in B95-8, 10% of cells in LCL-112, 5% of cells in Daudi and 3% of cells in EB2 and LCL-36. Lytically infected cells were recognized in addition by cytopathic effects visualized by staining with DAPI for evaluation by light microscopy. Cytopathic effects ranged from ballooning of the cell and moderate formation of chromatin clumps to holes and complete fragmentation of the nucleus (data not shown).
Correlation between Viral Protein Expression and DNA Signal Types 1–6
Latent membrane protein 1 was expressed in all cells documented to be latently infected and in some lytically infected cells. The proportion of latent membrane protein 1-positive cells in lymphoblastoid cell lines was about 85%, while in Burkitt's lymphoma cell lines the proportion varied between 4 and 7% (Namalwa, EB2 and Daudi) and up to 80% (Raji). Latent membrane protein 1 was diffusely distributed within the cytoplasm or formed patches at the cell periphery. Z fragment of Epstein–Barr replication activator and viral capsid antigen were expressed only in lytically infected cells. Latent membrane protein 1 positive cells were associated with signal types 1–5 (Figure 1h, l, m). Lytically infected cells were identified by the presence of diffuse nuclear staining for Z fragment of Epstein–Barr replication activator and signal types 4–6 (Figure 1o). Late stages of lytic viral infection were associated with viral capsid antigen expression and signal type 6 only (Figure 1r). Subsequently, the number of viral capsid antigen positive cells was smaller than the number of positive for Z fragment of Epstein–Barr replication activator. In some virus-producing cells, coexpression of latent membrane protein 1/Z fragment of Epstein–Barr replication activator (Figure 1p) and Z fragment of Epstein–Barr replication activator/viral capsid antigen were observed (Figure 1q). A coexpression of latent membrane protein 1/viral capsid antigen was not detected.
Determination of EBV Clonality by SB Analysis
The difference between episomal, integrated and linear EBV DNA was determined by band size and conformation of viral DNA by electrophoresis and Southern blot. Two DNA probes were used for Southern blot, corresponding to nucleic acid sequences adjacent to EBV DNA terminal repeats, fragments XhoIa and EcoRI1. Episomal DNA produced bands over 8 kilobases (kb) in size, which were of identical size using both probes. Raji, Daudi, EB2, all lymphoblastoid cell lines and B95-8 demonstrated comigration of bands when using both probes, thus demonstrating multiple episomal populations. Ladders below 8 kb were observed in Daudi, EB2, LCL-112, LCL-36 and B95-8, and likely represent linear extrachromosomal (replicating) DNA. Bands of different size were observed only in Namalwa, consistent with integration of viral DNA and the presence of variable junctions between viral and cellular DNA sequences (Figure 2).

Intracellular EBV genomic structures demonstrated by Southern blot analysis of BamHI digested cellular DNA probed with the XhoIa (left panel) and EcoRI1 (right panel) fragments, which represent unique cellular sequences flanking the right and left terminal repeats of the EBV genome. Integration and fusion of EBV DNA to cellular sequences are indicated by terminal fragments of different size, as in Namalwa (arrowheads). Possible integrated viral sequences are indicated by stars in the cell line Raji. Comigration of terminal fragments over 8 kb occurs as the result of fused terminal repeats representing episomes for the cell lines B95-8, LCL-MD, LCL-36, LCL-112, Raji, Daudi, and EB2. Terminal fragments of less than 8 kb represent linear genomes for the cell lines B95-8, LCL-112, Daudi and EB2. M=marker.
Discussion
Several efforts have been made to demonstrate the physical state of the EBV genome (linear, episomal or integrated) after infection into human cells.18, 33, 34, 35, 36, 37, 38, 39, 40 Linear EBV DNA circularizes into episomal form after infection.12, 41 Episomes are the EBV DNA form most often detected; however, integration of EBV DNA into the human genome has been recognized as an important mechanism for both the establishment of viral latency and for the occurrence of cellular transformation.18, 42, 43, 44 In this study, definition of latent and lytic intracellular EBV DNA forms by means of in situ hybridization has been demonstrated in characterized cell lines. We defined six signal types; large-disperse or type 1, small-punctate or type 2, combined or type 3, nuclear membrane associated or type 4, bubble or type 5, and spill-over or type 6 (Figure 3). Using light microscopy, viral protein expression and SB analysis in correlation with our in situ hybridization results, we demonstrated that these morphologies correspond to: episomal latent; integrated latent; simultaneous occurrence of integrated and episomal latent; switch from latency to viral replication; replicating viral DNA; release of virions from disrupted cells, respectively. Using in situ hybridization to recognize EBV DNA forms may prove to be important in investigating clinical material where no metaphase cells are available.

Schematic illustration summarizing the six different EBV ISH patterns observed in the latent and lytic pathway: ‘large-disperse’ type 1, represents an aggregation of episomes, ‘small-punctate’ type 2, represents stable integrated viral copies in doublets, ‘combined’ type 3, represents the combination of types 1 and 2, ‘nuclear membrane associated’ type 4, represents viral production and clustering of viral genomes near the nuclear membrane, ‘bubble’ type 5, represents the budding virus filling the entire cell and migrating to the cell membrane and finally ‘spill-over’ type 6, represents the virus in release. The expression pattern of the viral proteins latent membrane protein 1 (LMP1), Z fragment of Epstein–Barr replication activator (ZEBRA) and viral capsid antigen (VCA) associated with the six 6 EBV in situ hybridization patterns are shown.
Three forms of signal were seen in latent viral infection. The appearance of large-disperse forms was consistent with the fact that EBV episomes are large double-stranded, supercoiled DNA molecules able to form concatameric structures within the infected cell and able to undergo amplification.45, 46 Similarly, integrated DNA would appear as closely adjacent dots (doublets, or one signal per chromatid copy). As the Namalwa cell line contains only two integrated viral copies and completely lacks episomes, the distinct doublets seen in this cell line could only originate from integrated virus.17, 47 However, as single copies and small numbers of episomes could also result in doublet-like hybridization signals, the interpretation of individual punctate signals is not unequivocal. In the majority of latently infected cells, a combination of punctate doublets and large-disperse nuclear signals was observed, consistent with the presence of both episomal and integrated viral DNA.
In situ hybridization on metaphase chromosomes showed that integrated virus is located on specific chromosomal bands; it has been suggested that integrated viral genomes can induce chromosomal instability and result in breakpoints.48, 49 Whether EBV integration induces specific chromosomal abnormalities or oncogenic properties of genes in the vicinity associated with specific tumors, however, remains unclear. Our results support this view, but also indicate that in latently and lytically infected cell lines, viral integration is a considerably more common phenomenon than previously assumed.44, 50 Moreover, the presence of small numbers of episomal and chromosomally integrated EBV copies was observed in cells of newly transformed mantle cell lymphoma cell lines (Drs ME Williams and DG Bebb, personal communication), supporting the view that EBV integration mechanisms may be initiated shortly after infection. Latent membrane protein 1 is generally regarded to be a marker of latent viral infection,51 its expression was seen in all latently infected cell lines and was associated with EBV DNA signal types 1–3, confirming that these patterns were representative of latent infection.
Three lytic EBV DNA forms were identified by in situ hybridization. All three were detected in all cell lines analyzed with the exception of Namalwa, Raji and LCL-MD, which are known to harbor latent viral forms only. Lytic virus was present in more than 10% of cells of the producer cell line B95-8, and similar patterns were seen in the cell lines Daudi, EB2, LCL-112 and LCL-36, although in a considerably smaller cell fraction. These cells expressed the lytic EBV-specific proteins Z fragment of Epstein–Barr replication activator and viral capsid antigen and showed corresponding cytopathic features indicating production and release of EBV virions. The expression of Z fragment of Epstein–Barr replication activator in cells with nuclear membrane associated type 4 signals and no obvious signs of cytopathic effects likely reflected the switch from latent to lytic infection. Simultaneous detection of this protein and EBV DNA helped to rule out possible misinterpretation of nuclear membrane associated signals as a large-disperse component or combined signal type. The bubble signal type 5 in already ballooned nuclei represented the presence of lytic virus prior to release from the cell. High expression of viral capsid antigen in cells with spillover signal type 6 indicated that this hybridization pattern represents the latest stages of the lytic viral cycle, including incorporation of the viral genome into the capsid in preparation for release of mature viral particles from the cell. Some lytic cells were in mitosis (data not shown), in accordance with previous observations suggesting that EBV activation may be intimately linked with specific stages of the cell cycle.32, 52 Before, during and after release of EBV from lytically infected cells, it was impossible to discriminate between individual viral particles, but we estimated the number of viral copies to exceed several hundred per cell.
Latent membrane protein 1 was expressed in EBV-infected cells irrespective of the type of infection (latent or lytic), which is not surprising since latent membrane protein 1 is one of the few EBV genes expressed in both phases of the viral life cycle.53 Furthermore, individual latent membrane protein 1-positive cells within a cell line harbored more EBV DNA copies than latent membrane protein 1-negative cells and correlation between the level of latent membrane protein 1 expression and virus number or infection mode was observed (data not shown), in accordance with previous observations.54, 55 Discrimination of virus-producing and virus-nonproducing individual cells is, therefore, possible either by latent membrane protein 1/Z fragment of Epstein–Barr replication activator double labeling or by in situ hybridization alone.
Southern blot analysis using EBV probes for sequences adjacent to TR is the conventional approach to the determination of viral clonality and detection of the presence of episomal, integrated or linear infectious DNA forms.56, 57 We have applied this method to compare the sensitivity and specificity of the Southern blot and in situ hybridization approaches. Since Southern blot analysis probes for sequences adjacent to terminal repeats, this technique would not detect EBV integration via any segment of the viral genome other than terminal repeats. In the cell line Namalwa, which contains only integrated EBV, high molecular weight bands of different size corresponding to integration of the viral genome into different host sequences was observed. In cell lines with the combined in situ hybridization type 3 signal, comigrating SB bands of identical size were reliably detected. That we did not detect SB bands indicating viral integration in these cell lines can be explained by the presence of high copy numbers of episomal EBV obscuring detection of low copy numbers of integrated virus. Furthermore, the presence of up to 30 different chromosomal integration sites per cell line in combination with a dilution effect makes identification of single SB bands challenging.
The presence of replicative viral DNA seen in Southern blot correlated with the expression of Z fragment of Epstein–Barr replication activator and with increased occurrence of in situ hybridization signal types 4-6. The detection of Southern blot band ladders apparently depended on the proportion of lytic cells within the entire EBV-positive cell population. In the cell lines EB2 and LCL-36, therefore, which have a 3% lytic component, very weak bands were detected, whereas in Daudi, in which 5% of cells are lytic, a faint ladder array was seen. In both cell lines B95-8 and LCL-112, which have a lytic component over 10%, a distinct ladder array was observed.
As some viral proteins are present in both stages of the EBV life cycle and the SB approach cannot reliably detect low levels of integrated viral DNA in either latent or lytic stages, in situ hybridization provides a reliable, accurate and uncomplicated alternative technique. All forms of intracellular EBV DNA can be readily distinguished within individual cells by in situ hybridization, making this technique applicable to the study of EBV in single cells in clinical material. In this way, correlation between the presence of EBV DNA and stage of infection and other pathologic or clinical prognostic features may be investigated. For example, it has recently been observed that the cytotoxic agent hydroxyurea failed to inhibit the growth of cells harboring integrated EBV DNA, while cells harboring extrachromosomal EBV DNA were readily inhibited,58 so that discrimination between episomal and integrated latent EBV DNA forms becomes of particular clinical interest. Further studies using frozen and paraffin-embedded tissues are needed, however, to evaluate the practicality of this assay since it is possible that technical variables may inhibit its applicability.
References
Epstein MA, Achong BG . Various forms of Epstein–Barr virus infection in man: established facts and a general concept. Lancet 1973;2:836–839.
Epstein MA, Barr YM, Achong BG . A second virus-carrying tissue culture strain (Eb2) of lymphoblasts from Burkitt's lymphoma. Pathol Biol 1964;12:1233–1234.
Pagano JS . Viruses and lymphomas. N Engl J Med 2002;347:78–79.
Cohen JI . Epstein–Barr virus infection. N Engl J Med 2000;343:481–492.
Murray PG, Young LS . Themed issue: the biology and pathology of the Epstein–Barr virus. Mol Pathol 2000;53:219–221.
WHO-IARC. Epstein–Barr virus and Kaposi's sarcoma herpesvirus/human herpesvirus 8. Monographs on the Evaluation of Carcinogenic Risks to Humans, Vol. 70, 1998; http://www.iarc.fr/.
Kawanishi M . Epstein–Barr virus induces fragmentation of chromosomal DNA during lytic infection. J Virol 1993;67:7654–7658.
Alfieri C, Birkenbach M, Kieff E . Early events in Epstein–Barr virus infection of human B lymphocytes. Virology 1991;181:595–608.
Hurley EA, Thorley-Lawson DA . B cell activation and the establishment of Epstein–Barr virus latency. J Exp Med 1988;168:2059–2075.
Sugden B, Phelps M, Domoradzki J . Epstein–Barr virus DNA is amplified in transformed lymphocytes. J Virol 1979;31:590–595.
Mohyuddin A, Ayub Q, Siddiqi S, et al. Genetic instability in EBV-transformed lymphoblastoid cell lines. Biochim Biophys Acta 2004;1670:81–83.
Ohshima K, Suzumiya J, Kanda M, et al. Integrated and episomal forms of Epstein–Barr virus (EBV) in EBV associated disease. Cancer Lett 1998;122:43–50.
Lestou VS, Strehl S, Lion T, et al. High-resolution FISH of the entire integrated Epstein–Barr virus genome on extended human DNA. Cytogenet Cell Genet 1996; 74:211–217.
Lestou VS, De Braekeleer M, Strehl S, et al. Non-random integration of Epstein–Barr virus in lymphoblastoid cell lines. Genes Chromosomes Cancer 1993;8:38–48.
Delecluse HJ, Bartnizke S, Hammerschmidt W, et al. Episomal and integrated copies of Epstein–Barr virus coexist in Burkitt lymphoma cell lines. J Virol 1993;67:1292–1299.
Hurley EA, Agger S, McNeil JA, et al. When Epstein–Barr virus persistently infects B-cell lines, it frequently integrates. J Virol 1991;65:1245–1254.
Lawrence JB, Villnave CA, Singer RH . Sensitive, high-resolution chromatin and chromosome mapping in situ: presence and orientation of two closely integrated copies of EBV in a lymphoma line. Cell 1988;52:51–61.
Chang Y, Cheng SD, Tsai CH . Chromosomal integration of Epstein–Barr virus genomes in nasopharyngeal carcinoma cells. Head Neck 2002;24:143–150.
Glaser R, Tarr KL, Dangel AW . The transforming prototype of Epstein–Barr virus (B95-8) is also a lytic virus. Int J Cancer 1989;44:95–100.
Klein G, Dombos L, Gothoskar B . Sensitivity of Epstein–Barr virus (EBV) producer and non-producer human lymphoblastoid cell lines to superinfection with EB-virus. Int J Cancer 1972;10:44–57.
Zimmermann J, Hammerschmidt W . Structure and role of the terminal repeats of Epstein–Barr virus in processing and packaging of virion DNA. J Virol 1995;69:3147–3155.
Murray PG, Young LS . Epstein–Barr virus infection: basis of malignancy and potential for therapy. Exp Rev Mol Med 2001; http://www-ermm.cbcu.cam.ac.uk/01003842h.htm.
Bibeau F, Brousset P, Knecht H, et al. Epstein–Barr virus replication in Hodgkin disease. Bull Cancer 1994;81:114–118.
Raab-Traub N, Flynn K . The structure of the termini of the Epstein–Barr virus as a marker of clonal cellular proliferation. Cell 1986;47:883–889.
Cooper K . Physical state of human papillomavirus using non-isotopic in situ hybridisation. J Clin Pathol 1995;48:786.
Cooper K . Nonisotopic in situ hybridization in the detection of integrated HPV 16/18 in cervical cancers. Hum Pathol 1996;27:745.
Cooper K, Grayson W . Interpretation of the signal patterns produced by NISH in cervical neoplasia harbouring HPV. J Pathol 1997;183:126.
Cooper K, Herrington CS, Stickland JE, et al. Episomal and integrated human papillomavirus in cervical neoplasia shown by non-isotopic in situ hybridisation. J Clin Pathol 1991;44:990–996.
Henderson A, Ripley S, Heller M, et al. Chromosome site for Epstein–Barr virus DNA in a Burkitt tumor cell line and in lymphocytes growth-transformed in vitro. Proc Natl Acad Sci USA 1983;80:1987–1991.
Miller G . The switch between EBV latency and replication. Yale J Biol Med 1989;62:205–213.
Miller G . The switch between latency and replication of Epstein–Barr virus. J Infect Dis 1990;161:833–844.
Teo CG, Griffin BE . Visualization of single copies of the Epstein–Barr virus genome by in situ hybridization. Anal Biochem 1990;186:78–85.
Adams A, Lindahl T . Epstein–Barr virus genomes with properties of circular DNA molecules in carrier cells. Proc Natl Acad Sci USA 1975;72:1477–1481.
Andersson-Anvret M, Lindahl T . Integrated viral DNA sequences in Epstein–Barr virus-converted human lymphoma lines. J Virol 1978;25:710–718.
Boguszakova L, Hirsch I, Brichacek B, et al. Relationship between Epstein–Barr virus nuclear antigen and DNA genome number in superinfected and induced lymphoblastoid cell lines. J Gen Virol 1983;64:887–894.
Bornkamm GW, von Knebel-Doeberitz M, Lenoir GM . No evidence for differences in the Epstein–Barr virus genome carried in Burkitt lymphoma cells and nonmalignant lymphoblastoid cells from the same patients. Proc Natl Acad Sci USA 1984;81:4930–4934.
Coates PJ, Mak WP, Slavin G, et al. Detection of single copies of Epstein–Barr virus in paraffin wax sections by non-radioactive in situ hybridisation. J Clin Pathol 1991;44:487–491.
Szeles A . Fluorescence in situ hybridization (FISH) in the molecular cytogenetics of cancer. Acta Microbiol Immunol Hung 2002;49:69–80.
Szeles A, Falk KI, Imreh S, et al. Visualization of alternative Epstein–Barr virus expression programs by fluorescent in situ hybridization at the cell level. J Virol 1999;73:5064–5069.
Melcak I, Cermanova S, Jirsova K, et al. Nuclear pre-mRNA compartmentalization: trafficking of released transcripts to splicing factor reservoirs. Mol Biol Cell 2000;11:497–510.
Ohshima K, Suzumiya J, Ohga S, et al. Integrated Epstein–Barr virus (EBV) and chromosomal abnormality in chronic active EBV infection. Int J Cancer 1997;71:943–947.
Delecluse HJ, Bartnizke S, Hammerschmidt W, et al. Episomal and integrated copies of Epstein–Barr virus coexist in Burkitt lymphoma cell lines. J Virol 1993;67:1292–1299.
Becker Y, Shlomai J, Weinberg E, et al. Integration of Epstein–Barr virus DNA into cellular DNA of Burkitt lymphoblasts (Raji cell line). Isr J Med Sci 1974;10:1454–1457.
Gao Y, Lu YJ, Xue SA, et al. Hypothesis: a novel route for immortalization of epithelial cells by Epstein–Barr virus. Oncogene 2002;21:825–835.
Sato H, Takimoto T, Tanaka S, et al. Concatameric replication of Epstein–Barr virus: structure of the termini in virus-producer and newly transformed cell lines. J Virol 1990;64:5295–5300.
Corsini J, Cotmore SF, Tattersall P, et al. The left-end and right-end origins of minute virus of mice DNA differ in their capacity to direct episomal amplification and integration in vivo. Virology 2001;288:154–163.
Debiec-Rychter M, Croes R, De Vos R, et al. Complex genomic rearrangement of ALK loci associated with integrated human Epstein–Barr virus in a post-transplant myogenic liver tumor. Am J Pathol 2003;163:913–922.
Ohshima K, Suzumiya J, Ohga S, et al. Integrated Epstein–Barr virus (EBV) and chromosomal abnormality in chronic active EBV infection. Int J Cancer 1997;71:943–947.
Ohshima K, Kikuchi M, Masuda Y, et al. Epstein–Barr viral genomes in carcinoma metastatic to lymph nodes. Association with nasopharyngeal carcinoma. Acta Pathol Jpn 1991;41:437–443.
Jox A, Rohen C, Belge G, et al. Integration of Epstein–Barr virus in Burkitt's lymphoma cells leads to a region of enhanced chromosome instability. Ann Oncol 1997;8:131–135.
Yoshioka M, Kikuta H, Ishiguro N, et al. Latency pattern of Epstein–Barr virus and methylation status in Epstein–Barr virus-associated hemophagocytic syndrome. J Med Virol 2003;70:410–419.
Munch M . Epstein–Barr virus strain characterization. APMIS 1998;106:425–433.
Mann KP, Staunton D, Thorley-Lawson DA . Epstein–Barr virus-encoded protein found in plasma membranes of transformed cells. J Virol 1985;55:710–720.
Joske DJ, Emery-Goodman A, Bachmann E, et al. Epstein–Barr virus burden in Hodgkin's disease is related to latent membrane protein gene expression but not to active viral replication. Blood 1992;80:2610–2613.
Cordier-Bussat M, Calender A, Vuillaume M, et al. Expression of the Epstein–Barr virus (EBV) latent membrane protein is tightly regulated, independently of EB nuclear antigen 2 and of EBV integration or copy number. Virus Res 1993;27:55–69.
Gulley ML . Molecular diagnosis of Epstein–Barr virus-related diseases. J Mol Diagn 2001;3:1–10.
Gulley ML, Raphael M, Lutz CT, et al. Epstein–Barr virus integration in human lymphomas and lymphoid cell lines. Cancer 1992;70:185–191.
Mascolini M . HIV infection: old faithfuls and new frontiers. JIAPAC 1997; http://www.iapac.org/.
Acknowledgements
This work was supported by research funds of the Children's Institute of Cancer Research (CCRI) in St Anna Children's Hospital. GIGAX Foundation supported EEL. The publication costs and additional relevant work were supported by the Canadian Cancer Etiology Research Network/National Cancer Institute of Canada (Grant #010735).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Leenman, E., Panzer-Grümayer, R., Fischer, S. et al. Rapid determination of Epstein–Barr virus latent or lytic infection in single human cells using in situ hybridization. Mod Pathol 17, 1564–1572 (2004). https://doi.org/10.1038/modpathol.3800228
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800228
Keywords
This article is cited by
-
Accurate reconstruction of viral genomes in human cells from short reads using iterative refinement
BMC Genomics (2022)
-
Improved elongation factor-1 alpha-based vectors for stable high-level expression of heterologous proteins in Chinese hamster ovary cells
BMC Biotechnology (2014)
-
Base-pair resolution DNA methylome of the EBV-positive Endemic Burkitt lymphoma cell line DAUDI determined by SOLiD bisulfite-sequencing
Leukemia (2013)
