
Abstract
KIT is expressed not only in tumors derived from hematopoietic stem cells, melanocytes, germ cells, mast cells, and interstitial cells of Cajal, but also in other malignancies such as chromophobe renal cell carcinoma. This pattern of KIT expression prompted us to investigate the expression and mutation of c-kit gene exons 9, 11, 13, 17, and intron 17 in the different subtypes of renal cell carcinomas (n=66) and non-neoplastic kidneys (n=12). We found that KIT showed strong immunoreactivity in the cytoplasm of papillary renal cell carcinomas (100%), but on the cell membranes of chromophobe renal cell carcinomas (100%). Interestingly, a specific point mutation of the c-kit intron 17 (T->A) was found only in papillary renal cell carcinomas (94%). Our study demonstrates that the expression pattern and one mutation of c-kit may distinguish papillary renal cell carcinomas.
Similar content being viewed by others

Main
Renal cell carcinomas are increasingly common, constituting approximately 2% of all adult malignancies. The clinical outcome is poor and more than two-thirds of patients will die of their disease. Thus, it is very important to evaluate pathology specimens accurately for successful patient management.1 In addition, recent advances of molecular technology such as cDNA microarrays and proteomics may contribute to accurate diagnosis and discovery of new molecular targets of renal cell carcinomas. Renal cell carcinomas of the renal tubular epithelium are classified into conventional (clear cell), papillary, chromophobe, and collecting duct. The histopathologic classification of renal cell carcinomas is well correlated with distinct clinical features and cytogenetic abnormalities.2, 3 Thus, renal cell carcinomas are a heterogeneous group with distinct clinicopathologic features and cytogenetic abnormalities. In fact, based on the heterogeneity of renal cell cacinomas, gene expression profiling by cDNA microarrays has been compared among the subtypes of renal cell carcinomas.4, 5, 6 However, it is surprising that gene expression profiles in renal cell carcinomas are inconsistent in some aspects among the previous studies. In general, conventional renal cell carcinomas displayed overexpression of genes that are inducible by hypoxia. Chromophobe carcinomas showed upregulation of genes that are involved with oxidative phosphorylation and normally expressed by distal nephron. The c-kit is one of genes expressed in chromophobe renal cell carcinomas.5
The c-kit is the cellular homologue of the oncogene v-kit of HZ4 feline sarcoma virus. The c-kit proto-oncogene encodes a transmembrane receptor tyrosine kinase (KIT) in which the extracellular portion binds a ligand known as stem cell factor (SCF). Moreover, the SCF–KIT system plays an important role in the development and survival of hematopoietic stem cells, melanocytes, germ cells, mast cells, and interstitial cells of Cajal (ICC).7 KIT is not only expressed in tumors derived from these cell lineages, but also in other malignancies such as small cell lung cancer, pancreatic ductal carcinoma, angiomyolipoma, and chromophobe renal cell carcinoma.5, 8, 9, 10 Gain-of-function mutations of the c-kit gene are known in mast cell neoplasia and gastrointestinal stromal tumors.11, 12 The oncogenic mutations of c-kit gene or activation of KIT lead to activation of signal transduction cascades, which regulates cell proliferation, apoptosis, chemotaxis, and adhesion. Both wild and mutant KIT in gastrointestinal stromal tumors can be targeted by STI-571, also known as Gleevec.7 Interestingly, chromophobe renal cell carcinomas have been reported to express both KIT and its ligand, SCF.5 Although this study was carried out with a few limited cases of renal cell carcinomas, its results suggest that KIT signaling might have an important role in the oncogenesis of chromophobe renal cell carcinoma. In addition, KIT has been suggested to be a diagnostic marker and a therapeutic target for these tumors.5 This prompted us to investigate the expression and mutation of c-kit gene in the different subtypes of renal cell carcinomas. Thus, to determine whether c-kit is expressed or its gene is mutated in renal cell carcinomas, we studied 66 cases of renal cell carcinomas and 12 cases of adjacent non-neoplastic kidneys. Immunohistochemistry for c-kit, polymerase chain reactions (PCR) and direct DNA sequencing for c-kit exons 9, 11, 13, 17, and intron 17 were performed.
Materials and methods
Tissue Samples
We analyzed 66 cases of renal cell carcinoma, including the conventional (n=28), papillary (n=18), and chromophobe type (n=20) and 12 cases of the corresponding non-neoplastic kidney obtained from the Department of Pathology, Korea University Medical Center. The non-neoplastic kidneys were obtained from the tissue sections adjacent to the corresponding renal cell carcinomas (six cases of conventional, three cases of papillary, and three cases of chromophobe renal cell carcinomas). All specimens were fixed in 10% buffered formalin, embedded in paraffin, and stained with hematoxylin and eosin, and Hale's colloidal iron. The classification, nuclear grade and tumor stage of the renal cell carcinomas were determined according to the criteria proposed by Fuhrman et al, and WHO.13, 14 Papillary renal cell carcinomas were also subdivided into types I (n=7), and II (n=11).15
Immunohistochemistry
Immunohistochemistry was performed using a ChemMate EnVision Detection Kit (DAKO, Carpinteria, CA, USA) to eliminate nonspecific immunoreactions by endogenous biotin in the kidney. Blocking peptide for anti-KIT antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) was also used to ensure the specificity of KIT immunostaining. For immunohistochemistry, tissue sections were deparaffinized and rehydrated. Sections placed in 10 mM EDTA (pH 8.0) were heated in a microwave for 5 min 30 s. After incubation with 0.3% H2O2 for 20 min, slides were incubated with primary polyclonal rabbit anti-human KIT [1:100 and 1:250(v/v)] (DAKO, Carpinteria, CA, USA) at room temperature (RT) for 1 h.16, 17 After incubation with secondary antibody at RT for 30 min, the sections were developed with 3,3′-diaminobenzidine (DAB) and counterstained with Mayor's hematoxylin. The blocking peptides and positive (cases of gastrointestinal stromal tumors) and negative controls (omission of primary antibody) were used to determine the specificity of KIT immunostaining.16 Pretreated slides as mentioned above were incubated with combined antibody (primary KIT antibody: blocking peptides=1:5) at RT for 2 h. Slides were then stained using the same protocol.
PCR and Direct DNA Sequencing
DNA was extracted from formalin-fixed, paraffin-embedded tissues using a High Pure Template DNA Isolation Kit (Roche Diagnostics GmbH, Mannheim, Germany). Somatic mutations in exons 9, 11, 13, 17, and intron 17 of the c-kit gene were amplified by PCR using the following oligonucleotide primer pairs:18, 19 for exon 9, 5′-TCC TAG AGT AAG CCA GGG CTT-3′/5′-TGG TAG ACA GAG CCT AAA CAT CC-3′; for exon 11, 5′-CCA GAG TGC TCT AAT GAC TG-3′/5′-AGC CCC TGT TTC ATA CTG AC-3′; for exon 13, 5′-GCT TGA CAT CAG TTT GCC AG-3′/5′-AAA GGC AGC TTG GAC ACG GCT TTA-3′; and for exon 17 and intron 17; 5′-CCT CCA ACC TAA TAG TGT AT-3′/5′-TCT GGC TAG ACC AAA ATC AC-3′ and 5′-CAT GGT CGG ATC ACA AAG AT-3′/5′-ATT ATG AAA GTC ACG GAA AC-3′. PCR was performed in a mixture of 25 μl containing 150 ng of sample DNA; 10 × PCR buffer; 0.2 mM dNTP; 2 pmol/μl primers; and 1 U of Taq DNA polymerase. After denaturation at 95°C for 5 min, DNA amplification was performed over 40 cycles consisting of denaturation at 94°C for 45 s, annealing at 55–60°C for 45 s, and extension at 72°C for 45 s. PCR products were separated in 2.0% agarose gels. The PCR products were purified and sequenced using an ABI prism 310 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA)
Results
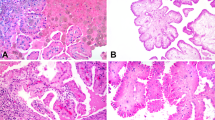
Interestingly, KIT expression was observed in all cases of papillary and chromophobe renal cell carcinomas, but not in conventional renal cell carcinomas. The immunostaining pattern of KIT was very different in the papillary and chromophobe types. KIT was expressed in the cytoplasm of papillary carcinomas, whereas it was expressed on the cell membrane of chromophobe carcinomas (Figure 1c and e). There was no variation in KIT expression between types I and II of papillary renal cell carcinomas. KIT expression was not correlated with Fuhrman grade or tumor stage in these carcinomas (Table 1). The possibility of nonspecific immunoreactions for KIT was excluded by the results of experiments using blocking peptides for anti-KIT antibody, positive and negative controls, and diluted primary antibody at 1:250 (v/v) (Figure 1d and f). In addition, mast cells showing strong membranous immunoreactivity were used as an internal control for KIT expression. KIT was also expressed in the tubular cells of non-neoplastic kidneys, where it was localized in the proximal and distal tubular cells, but not in the glomerular cells (Figure 1a). It was noteworthy that the immunostaining patterns of KIT in non-neoplastic kidneys were reminiscent of those in renal cell carcinomas.

Immunohistochemistry of KIT. (a) KIT was expressed in the proximal and distal tubular cells of non-neoplastic kidneys. (b) KIT was not expressed in conventional renal cell carcinomas. (c) KIT was strongly expressed in the cytoplasm of papillary renal cell carcinomas. (d) After treating with combined anti-KIT antibodies and blocking peptides, KIT expression was blocked in papillary renal cell carcinomas. (e) KIT was strongly expressed on the cell membrane of chromophobe renal cell carcinomas. (f) After treating with combined anti-KIT antibodies and blocking peptides, KIT expression was also blocked in chromophobe renal cell carcinomas (immunoperoxidase, × 200 magnification).
All samples were subjected to mutational analyses of c-kit gene. No mutation in exons 9, 11, 13, or 17 was observed in any types of renal cell carcinoma or in non-neoplastic kidneys. However, it was noteworthy that a point mutation in c-kit intron 17 was found only in papillary renal cell carcinomas, but not in conventional renal cell carcinomas, chromophobe renal cell carcinomas, or non-neoplastic normal kidneys (Figure 2). This mutation, which has not been previously reported, was specifically found in 17 (94%) of 18 papillary renal cell carcinomas. The point mutation (T->A) was located on the 43rd base of the c-kit intron17, which is away from the exon 17-intron 17 border (Figure 2b, d, and f). One case of type II papillary renal cell carcinomas did not show the mutation of c-kit intron 17.

Mutational analysis of c-kit intron 17 by polymerase chain reactions-direct DNA sequencing. A heterozygous point mutation (T->A) of c-kit intron 17 was seen only in papillary renal cell carcinomas (b, d, and f). In contrast, no mutation of c-kit was found in (a) non-neoplastic kidneys, (c) conventional renal cell carcinomas, and (e) chromophobe renal cell carcinomas.
Discussion
To our knowledge, we found for the first time that papillary renal cell carcinoma shows KIT expression in the cytoplasm of tumor cells and a specific point mutation of c-kit intron 17. In addition, chromophobe renal cell carcinoma showed KIT expression on the tumor cell membrane without any mutation of c-kit. Similarly, KIT was expressed in the cytoplasm or on the cell membrane of the proximal and distal tubular cells of the non-neoplastic kidneys. KIT expression in renal cell carcinomas was well correlated with histological subtype, but independent of clinical stage or pathologic grade.
KIT was expressed in the cytoplasm of papillary renal cell carcinomas and on the cell membrane of chromophobe renal cell carcinomas. However, KIT was not expressed in papillary urothelial carcinomas arising from the renal pelvis (data not shown). Our results of chromophobe carcinoma are fully in agreement with those of the previous study by Yamazaki et al.5 In case of papillary renal cell carcinomas, KIT expression, and mutational analysis of c-kit were not mentioned in detail in Yamazaki's study. Although it was presented only in a figure of their paper, two cases of papillary renal cell carcinomas did not express mRNA of c-kit but one case did mRNA of stem cell factor.5 In our study, the possibility of nonspecific immunoreactions for KIT was excluded by repeated experiments using blocking peptides for anti-KIT antibody, positive and negative controls, and diluted primary antibody. Since KIT is a receptor tyrosine kinase located on the cell membrane, it is believed that the immunostaining pattern of KIT would be membranous. However, papillary renal cell carcinomas showed diffuse cytoplasmic pattern with occasional cytoplasmic punctuate staining. It has been suggested that this immunostaining pattern may reflect the different type of c-kit mutations, because most gastrointestinal stromal tumors show unequivocal diffuse, strong cytoplasmic positivity.20 This hypothesis is also supported by the fact that chromophobe renal cell carcinomas, showing no mutation in c-kit, exhibit distinct membranous KIT immunostaining pattern. The membranous expression of chromophobe renal cell carcinomas is in contrast to that shown by papillary renal cell carcinomas. Papillary renal cell carcinomas showed a heterozygous, point mutation of c-kit intron 17, which is rather far from the splicing site of exon 17. This mutation of c-kit intron 17 was not found in any non-neoplastic kidneys. Furthermore, this mutation was not shown by any other known single-nucleotide polymorphisms in GenBank (National Center for Biotechnology Information, NIH, Bethesda, MD, USA). Since this mutation is not on or near the splicing site of c-kit exon17, it is less likely to produce a KIT isoform by an alternative splicing. It is, however, speculated that the novel mutation of the c-kit intron 17 adjacent to exon17, encoding the tyrosine kinase domain, might affect the enzymatic activity of KIT.7 In addition, the expression pattern and the mutation of the c-kit intron may help distinguishing papillary renal carcinomas from other subtypes of renal cell carcinomas, containing papillary structures, and from papillary urothelial carcinomas in the renal pelvis.
In conclusion, papillary renal cell carcinomas were found to show strong cytoplasmic KIT expression and a specific point mutation of c-kit intron 17 (T->A). Thus, the expression pattern and the mutation of the c-kit intron may be helpful to distinguish papillary carcinomas from other subtypes of renal cell carcinomas or from papillary urothelial carcinoma of the renal pelvis. It is possible that KIT plays a role in the oncogenesis of papillary renal cell carcinoma, although its significance needs to be further investigated.
References
Fleming S, O'Donnell M . Surgical pathology of renal epithelial neoplasms: recent advances and current status. Histopathology 2000;36:195–202.
Storkel S, Eble JN, Adlakha K, et al. Classification of renal cell carcinoma: Workgroup No. 1. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer 1997;80:987–989.
Cheville JC, Lohse CM, Zincke H, et al. Comparisons of outcome and prognostic features among histologic subtypes of renal cell carcinoma. Am J Surg Pathol 2003;27:612–624.
Young AN, de Oliveira Salles PG, Lim SD, et al. Beta defensin-1, parvalbumin, and vimentin: a panel of diagnostic immunohistochemical markers for renal tumors derived from gene expression profiling studies using cDNA microarrays. Am J Surg Pathol 2003;27:199–205.
Yamazaki K, Sakamoto M, Ohta T, et al. Overexpression of KIT in chromophobe renal cell carcinoma. Oncogene 2003;22:847–852.
Takahashi M, Sugimura J, Yang X, et al. Gene expression profiling of renal cell carcinoma and its implications in diagnosis, prognosis, and therapeutics. Adv Cancer Res 2003;89:157–181.
Heinrich MC, Rubin BP, Longley BJ, et al. Biology and genetic aspects of gastrointestinal stromal tumors: KIT activation and cytogenetic alterations. Hum Pathol 2002;33:484–495.
Sekido Y, Obata Y, Ueda R, et al. Preferential expression of c-kit protooncogene transcripts in small cell lung cancer. Cancer Res 1991;51:2416–2419.
Nio Y, Omori H, Toga T, et al. Immunohistochemical expression of receptor-tyrosine kinase c-kit protein in invasive ductal carcinoma of the pancreas. Anticancer Drugs 2003;14:313–319.
Makhlouf HR, Remotti HE, Ishak KG . Expression of KIT (CD117) in angiomyolipoma. Am J Surg Pathol 2002;26:493–497.
Furitsu T, Tsujimura T, Tono T, et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest 1993;92:1736–1744.
Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577–580.
Fuhrman SA, Lasky LC, Limas C . Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol 1982;6:655–663.
Guinan P, Sobin LH, Algaba F, et al. TNM staging of renal cell carcinoma: Workgroup No. 3. Union International Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer 1997;80:992–993.
Delahunt B, Eble JN . Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol 1997;10:537–544.
Hornick JL, Fletcher CD . Validating immunohistochemical staining for KIT (CD117). Am J Clin Pathol 2003;119:325–327.
Lucas DR, al-Abbadi M, Tabaczka P, et al. c-Kit expression in desmoid fibromatosis. Comparative immunohistochemical evaluation of two commercial antibodies. Am J Clin Pathol 2003;119:339–345.
Lasota J, Wozniak A, Sarlomo-Rikala M, et al. Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. A study of 200 cases. Am J Pathol 2000;157:1091–1095.
Taniguchi M, Nishida T, Hirota S, et al. Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Res 1999;9:4297–4300.
Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol 2002;33:459–465.
Acknowledgements
We thank Sang-Ju Lee, Mi-Ran Jeong, Eun-Ja Lee, and Sung-Su Lee for their excellent technical assistance. The Brain Korea 21 Task Force and the Korea Lung Tissue Bank supported the study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lin, ZH., Han, E., Lee, E. et al. A distinct expression pattern and point mutation of c-kit in papillary renal cell carcinomas. Mod Pathol 17, 611–616 (2004). https://doi.org/10.1038/modpathol.3800108
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800108
Keywords
This article is cited by
-
Tr-KIT/c-KIT ratio in renal cell carcinoma
Molecular Biology Reports (2019)
-
Inzidenz und Langzeitprognose des papillären Nierenzellkarzinoms
Der Urologe (2011)
-
Similar developmental patterns in immunolocalisation of stem cell factor and KIT in bovine meso- and metanephros
Histochemistry and Cell Biology (2010)
-
Chromophobe renal cell cancer - review of the literature and potential methods of treating metastatic disease
Journal of Experimental & Clinical Cancer Research (2009)
-
Incidence and long-term prognosis of papillary renal cell carcinoma
Journal of Cancer Research and Clinical Oncology (2009)
