
Abstract
Adenocarcinomas of the lung are characterized by morphological heterogeneity, and since carcinogenesis has been suggested to be a multistep process involving sequential accumulation of multiple genetic alterations, the morphological heterogeneity may represent a cross-sectional view of genetic alterations within individual tumors. Therefore, to elucidate whether, and which, genetic alterations accumulated in relation to morphological cancer progression, we examined 56 microdissected sites for topographical distribution of loss of heterozygosity (LOH) in 12 adenocarcinomas of the lung with bronchioloalveolar (BA) and invasive components in their primary tumors and metastases to lymph nodes. The morphological changes from noninvasive BA lesions to invasive and metastatic components were characterized by a significant rise in the prevalence of allelic losses (P<0.05). Individually, eight cases (67%) showed accumulation of genetic alterations from BA lesions to metastases. LOHs in multiple foci in one case were compared to determine whether they were shared at all tumor sites as an early event or localized in metastases as an additional event. LOHs at 5q and 17p may be crucial steps in the early phase of development to metastasis, while 18q loss may be an additional step. These findings suggested that the cancer cells in some pulmonary adenocarcinomas evolved from the BA lesions to the invasive and metastatic lesions.
Similar content being viewed by others

Main
Carcinoma of the lung is one of the most common human cancers and is the predominant cause of cancer-related death throughout the world. Of the four major histological subtypes of lung cancer, the incidence of adenocarcinoma has been gradually increasing, but the morphological and genetic progression schemes of adenocarcinoma of the lung have not been elucidated as clearly as those of colon cancer,1 which is the human cancer that has been most intensively investigated in this regard. Elucidation of the genetic sequence responsible for the progression of adenocarcinoma of the lung from in situ to invasive and metastatic carcinoma is an important goal in understanding the biological basis of this malignancy.
One of the most characteristic features of adenocarcinoma of the lung is its high degree of morphological heterogeneity. We have often observed morphologically noninvasive bronchioloalveolar carcinoma (BAC) components replacing pre-existing alveolar epithelium (the replacing-growth-type) at the periphery of invasive adenocarcinomas. According to the WHO classification,2 they were diagnosed as ‘adenocarcinoma mixed bronchioloalveolar and other subtypes (acinar, solid, papillary).’ This morphological heterogeneity may represent a cross-sectional view of clonal evolution within individual tumors. That is, some noninvasive BACs may evolve into invasive adenocarcinoma as they acquire invasiveness during carcinogenesis, as observed in the adenoma–carcinoma sequence of colon carcinogenesis.1, 3, 4, 5 Adenocarcinomas mixed bronchioloalveolar (BA) and other subtypes are supposed to contain bronchioloalveolar (BA) components of BACs and invasive components evolving from the BAC in the individual tumor. They contain noninvasive components and invasive components that we usually observe in each tumor as part of the adenoma–carcinoma sequence. In addition, it has never been determined whether adenocarcinomas containing a morphologically noninvasive BA component (adenocarcinoma mixed BA and other subtypes) evolve from BAC. We recently examined 66 cases of replacing-growth-type small adenocarcinoma of the lung less than 2 cm in size to investigate allelic losses at eight loci on the eight chromosomes carrying the most important cancer-associated genes by the laser capture microdissection method, in which cancer cells are selectively collected.6 The 66 cases were divided into three groups according to the Noguchi's classification of small adenocarcinomas of the lung:7 12 localized bronchioloalveolar carcinomas (LBACs), 28 LBACs with alveolar collapse, and 26 LBACs with active fibroblastic proliferation. We confirmed that the prevalence of loss of heterozygosity (LOH) increased as adenocarcinoma of the lung underwent histological progression from LBAC (16.7%) to small but advanced LBAC with fibroblastic proliferation (96.2%). Deletions of four of the markers, 3p, 17p, 18q, and 22q, increased significantly during the carcinogenic steps from noninvasive to invasive carcinoma, and these molecular genetic data were consistent with the morphological progression of the adenocarcinoma.
It has been suggested that certain types of genetic alterations may be involved in the early phase of tumorigenesis, whereas others may play a role in late events during tumor progression. However, such inferences have essentially been based on statistical arguments after analysis of a set of different tumors6, 8, 9 and whether they are indeed applicable to the progression scheme in individual tumors has not been specifically addressed in regard to adenocarcinoma of the lung. An alternative approach therefore seemed to be necessary, for those relying on the statistical correlation between the frequency of each genetic alteration and histological and/or disease progression, but such an approach has rarely been taken in studies of the genetic changes in adenocarcinoma of the lung.
The pathologic staging of malignant tumors, including of non-small cell lung carcinoma (NSCLC), is based on the concept that primary tumor growth and local invasion precede lymphatic dissemination, which is regarded as a later event in lung cancer progression. From the standpoint of molecular genetics as well, the metastatic event is considered to be a relatively late event that follows multiple sequential and selective steps of clonal evolution. Metastases are thought to be established through selected clonal tumor cells that carry all genetic alterations involved in the genesis and progression of carcinoma.10, 11 However, no studies have carefully compared the accumulation of genetic changes and histopathological progression from a noninvasive lesion to an invasive or metastatic lesion in an individual pulmonary adenocarcinoma mixed BA and other subtypes.
In the present study, we examined the topographical distribution of LOH events occurring in the process of neoplastic progression within individual tumors and assessed whether previous inferences are indeed applicable to the progression scheme within individual tumors. To do so, we examined multiple pathologically well-defined specimens from individual tumors with lymph node metastases in order to investigate whether, and which genetic alterations are accumulated in relation to histopathological progression in individual tumors.
Materials and methods
Patients and Tissue Samples
We selected 12 cases of adenocarcinoma of the lung in which the primary tumor measured 3 cm or less in greatest dimension and contained BA components and invasive components (adenocarcinoma mixed BA and other subtypes) and there were metastases in the resected lymph nodes. All tumor samples were obtained from surgical resections at the Department of Thoracic Oncology of the National Cancer Center Hospital East (Chiba, Japan) between 1998 and 2000. All patients underwent lobectomies and lymph node dissections curative intent with no preoperative adjuvant therapy.
A 3-μm-thick section from each block was stained with hematoxylin–eosin (HE) and used as a guide to localize regions of interest for microdissection. We examined all identifiable components that appeared histotopographically different and contained sufficient cells (more than 100 cells). To investigate the clear relationship between pathology and genetic alteration, we collected the lesions whose subtypes we could define clearly and which consisted of the histologically uniform cells. In the microdissection analysis, two or three 10-μm-thick sections from each specimen were deparaffinized with xylene and stained with hematoxylin alone. The stained sections were dried, and multiple lesions in each tumor were microdissected separately with a Pixcell Laser Capture Microdissection System (Arcturus Engineering Inc., Mountainview, CA, USA).12 Finally, 100–200 tumor cells and the same number of normal cells were microdissected from each specimen, and their genomic DNAs were extracted as described previously. Separated tumor areas (3–6 areas per case) that were histotopographically dissimilar were selected microscopically on HE-stained sections. We identified a total of 56 foci, which included samples from the 12 BA component sites, 30 invasive regions of primary tumors (17 acinar, eight solid, five papillary), and 14 metastases (13 lymph node metastases, one intrapulmonary metastasis). Normal control DNAs were extracted from the corresponding lymph nodes without metastasis.
Multiplex Polymerase Chain Reaction–Loss of Heterozygosity Analysis
To evaluate LOH, we used 24 polymorphic microsatellite markers located at the following sites in the 10 genes reported to play a major role in human carcinogenesis:13, 14, 15, 16, 17, 18, 19, 20, 21, 22 FHIT (3p) (D3S1300, D3S1312, and D3S1313), APC (5q) (D5S346 and D5S82), p16 (9p) (D9S171 and D9S162), TSC-1 (9q) (D9S149, D9S150 and DBH), Int-2 (11q) (INT-2), Rb (13q) (D13S270, D13S273, and D13S176), TSC-2 (16p) (D16S291 and D16S292), p53 (17p) (TP53 and D17S520), Smad 4 (18q) (D18S46, D18S363, and D18S474), and Band M (22q) (D22S1140, D22S1170, and D22S1161). The use of more than one microsatellite marker ensured a higher yield of information for each genomic locus. Polymerase chain reaction (PCR) reactions for the fluorescent-labeled markers were carried out in a volume of 20μl that included 2 or 3 μl of 10 × PCR buffer and 5–25 pmol of each primer, 1 μl of template DNA, 200 μM of each deoxynucleotide triphosphate (dNTP), and 1.0 or 1.5 U of Taq DNA polymerase. To detect the amplified fragments, the samples were run on a Model 377 Genetic Analysis System (Applied Biosystems, Foster City, CA, USA) using Gene Scan 377 software (Applied Biosystems, Foster City, CA, USA). Markers that identified two distinguishable alleles of different sizes but similar intensity in normal DNA were termed ‘informative’ (heterozygous). Markers that yielded a single major peak in normal DNA were termed ‘noninformative’ (homozygous). LOH was defined as loss of one allele that corresponded to an allele present in informative cases. A result was scored as LOH if there was a greater than 50% reduction in allele ratio in the tumor relative to the normal control DNA.
DNA Analysis
We used two approaches to data analysis:9 (1) to correlate morphologic changes with allelic losses, we calculated the mean Fractional Regional Loss (FRL) index, defined as: FRL index=total number of chromosomal regions with LOH/total number of informative regions, and (2) to determine whether the deletions in the individual chromosomal regions were progressive in individual foci, we determined the frequencies of loss of individual markers on the basis of a Fractional Allelic Loss (FAL) index defined as: FAL index=total number of markers with LOH/total number of informative markers. Fisher's exact test was used for the statistical analysis in these approaches, and probability values of P<0.05 were regarded as statistically significant.
Results
Accumulation of Genetic Alterations in Individual Tumors
Microsatellite markers on 10 chromosome arms were used to perform a PCR-based multifocal LOH assay on a total 56 tumor sites microdissected from 12 adenocarcinomas mixed BA and other subtypes of the lung. The results for each locus are summarized in Table 1. At least one allelic loss was detected in all 56 sites examined. To gain a greater understanding of the relationship between the morphologic and genetic anatomy of each specimen, two representative cases are shown in Figures 1 and 2, respectively. Normal components are designated ‘N,’ bronchioloalveolar components ‘BA,’ acinar components ‘A,’ solid components ‘S,’ papillary components ‘P,’ intrapulmonary metastases ‘PM,’ and lymph node metastases ‘LNM.’ Multiple foci representing the same histology are distinguished by a numerical designation. We used the changes (including the presence of allelic loss and the specific allele lost) to determine whether the foci in individual cases were clonally related. A diagram shows the least complex pathway of tumor progression compatible with the LOH data in each case. The diagrams were drawn based on the assumption that, whenever compatible with the data, an observed LOH event represents a single occurrence in the evolution of the tumor, so that tumor components with LOH at the same locus are postulated to share a common precursor.

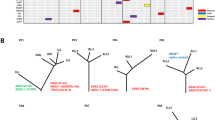
Analysis of LOH in an adenocarcinoma mixed BA, acinar, and solid (case 2). (a), Histology: low-power view showing regions chosen for laser capture microdissection (hematoxylin and eosin staining). BA: bronchioloalveolar component; S1 and S2: solid component dissected from different regions of the specimen; A1 and A2: acinar component dissected from different regions of the specimen; LNM (A): lymph node metastasis (acinar component). Primary tumor, which included a focus of BA-type carcinoma (BA) and two invasive acinar carcinomas (A1 and A2) and two invasive solid carcinomas (S1 and S2), and a metastatic tumor (lymph node metastasis; LNM) were analyzed. The metastatic tumor was acinar carcinoma. (b) Microscopic appearance of the four tumor areas (HE staining; original magnification, × 200). b1: BA component; b2: solid component; b3: acinar component; b4: acinar component in the lymph node metastasis. (c) LOH data obtained from microdissected tumor components. □: LOH of top band, ▪: LOH of bottom band, -: no LOH, blank space: noninformative (d) Clonal relationship between microdissected tumor components suggested by LOH data. Open circles indicate a putative intermediate or precursor cell. LOH is indicated on the diagrams by microsatellite markers that showed allelic loss. BA, which contained one allelic loss (marker D22S1140), may be the precursor lesion of other invasive lesions as well as metastatic lesion. In the same manner, S1, which contained one allelic loss (marker TP53) in addition to the loss (marker D22S1140), may be the precursor lesion of other invasive and metastatic lesions. A1, which contained six additional allelic losses, may be the precursor lesion of the remaining lesions. Three lesions (S2, A2, and LNM (A)) had lost alleles at all 10 markers.

Analysis of LOH in an adenocarcinoma mixed BA and acinar (case 3). (a) Histology: BA: BA component; A1, A2, and A3: acinar component dissected from different regions of the specimen; LNM (A): lymph node metastasis (acinar component). Tumor components included BA-type carcinoma (BA) and three foci of invasive acinar components (A1–3) in the primary tumor and another acinar focus in the lymph node metastasis (LNM (A)). (b) LOH data obtained from microdissected tumor components. □: LOH of top band, ▪: LOH of bottom band, -: no LOH, blank space: noninformative. (c) Clonal relationship between microdissected tumor components suggested by LOH data. Designation of LOH results is as in Figure 1. All components contained two allelic losses (marker D5S82 and D16S291). A1 showed LOH at the smaller allele of TP53 that was not detected in other components. In the same manner, only the BA component contained one allelic loss (marker D22S1140). These results suggest that the A1 and BA components must have evolved along subclonal pathways and that they shared common precursors, indicated by the open circle in the diagram. A2 showed LOH at the larger allele of TP53 in addition to the common precursor, and may be the precursor lesion of A3 and LNM (A).
In case 2 (Figure 1), genetic alterations accumulated in the evolution of the tumor from the BA component to the invasive and metastatic tumor components. Case 3 (Figure 2), on the other hand, is characterized by alterations in the primary tumor that were not detectable in the metastasis (TP53 (smaller allele) in A1, D22S1140 in BA). Although genetic alterations accumulated in the evolution of the tumor to metastasis, two subclonal pathways were demonstrated in the primary tumor.
As shown in these representative cases, genetic alterations accumulated during the evolution of the tumors. However, as demonstrated by case 3, tumor evolution along a single clonal pathway from BA to invasive and metastatic components was not detected in every case. Four of the BA components (4/12: 33.3%) and five of the invasive components (5/30: 16.7%) appeared to have evolved through independent subclonal pathways. In regard to the relationship among histological subtypes in the invasive lesions, in seven cases there were multiple morphological subtypes in the primary tumors, even though two of them displayed an identical LOH pattern. The remaining five cases did not show any distinct relationship between morphology and LOH status.
Allelic Loss in Histopathological Progression
(Figure 3) To elucidate whether accumulation of genetic alterations is related to histopathological progression, we compared mean FRL indices among BA-type lesions, invasive lesions, and metastases. The mean FRL indices for invasive lesions of the primary tumor and metastasis (0.66 and 0.69) were significantly higher than that for BA-type lesion (0.55); (P<0.05), however, the difference between the mean FRL index of the invasive components and metastatic components was not significant. The histological change from BA to invasive and to metastatic components was characterized by a significant rise in the mean FRL indices. The 12 cases could be divided into three groups according to the patterns of allelic losses. (1) Alleles that were retained in the BA lesion were deleted in either metastatic lesions or invasive and metastatic lesions. Eight cases (67%) showed accumulation of genetic alterations from BA lesions to metastases. (2) All foci had the same allelic losses. Two cases (17%) showed the same LOH patterns in all foci examined. (3) The markers used in this study could not be used to analyze the LOH patterns. Two cases, cases 5 and 6, had allelic loss in only one region in only one component (invasive and BA lesion), and the other components showed the same LOH patterns.

Mean fractional regional loss indices for the BA, invasive, and metastatic components in 10 chromosomal regions.
Prevalence and Intratumor Topography of LOH Events
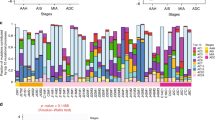
LOH was observed in 75.0% (126/168) of the informative markers on the 10 chromosome arms, including 3p (77.3%), 5q (80.0%), 9p (88.9%), 9q (85.2%), 11q (37.5%), 13q (83.3%), 16p (28.6%), 17p (93.8%), 18q (91.7%), and 22q (46.7%). In order to investigate the sequence of molecular genetic changes involved in the development of metastasizing adenocarcinoma mixed BA and other subtypes of the lung, we analyzed the topographical distribution of allelic losses in the tumors. Multiple foci from the same case were compared to determine whether the LOH events were shared at all tumor sites as a relatively early event derived from a progenitor cell, or localized in either metastatic lesions or invasive and metastatic lesions as an additional event directing toward invasion and metastasis. Losses on 5q were detected uniformly in the various regions of cases that tested positive for LOH (two cases were uniformly negative and three were noninformative), while homogeneous distribution of LOH was observed on 3p (70.6%), 9p (25.0%), 9q (43.5%), 11q (0.0%), 13q (66.7%), 16p (50.0%), 17p (80.0%), 18q (59.1%), and 22q (71.4%; and Figure 4). Losses on 11q were not detected uniformly at all tumor sites tested, but localized in either metastatic lesions or invasive and metastatic lesions, while heterogeneous distribution of LOH was observed on 3p (11.8%), 9p (37.5%), 9q (39.1%), 13q (33.3%), 16p (50.0%), 17p (13.3%), 18q (40.9%), and 22q (14.3%; and Figure 4). As shown in Figure 4, 5q loss was observed frequently (80.0%) and most uniformly (100%) in multiple sites, and was therefore interpreted as an obligatory early event in the progression of metastasizing pulmonary adenocarcinoma mixed BA and other subtypes. As 17p loss was most frequent (93.3%) and relatively uniform (80.0%), it was also interpreted as an early event. The loss of 18q was relatively frequent (91.7%) and heterogeneous (40.9%), and therefore indicative of late alterations required for malignant progression.

Prevalence and intratumor heterogeneity of allelic loss in primary tumors and lymph node metastases. PT, primary tumor.
Discussion
Invasive and/or metastatic components contained more additional allelic losses than BA components. It has already been postulated that tumor cells evolved from BA to invasive and metastatic components because of the difference in histology between the lepidic pattern along the alveolar walls in BA components and invasiveness in other components.7 Almost all metastases contained the same or more allelic losses when compared with invasive lesions in each individual tumor, although no significant difference was found in the mean FRL indices of the invasive and metastatic lesions. This suggests that tumor cells that showed noninvasive BA-type morphology evolved into invasive lesions and then to metastases, acquiring the invasive and metastatic phenotype through the process of clonal evolution occurring during multistep tumor progression. Eight cases (67%) actually showed accumulation of genetic alterations during morphological progression, but the tumor components examined thus far in the remaining four cases have shown genetic homogeneity or no obvious accumulation of genetic alterations despite great morphological divergence. This might be explained by the genetic alterations being present on loci other than those examined in this study. Although the additional allelic losses found in the metastases can be explained by the accumulation of genetic aberrations during the course of tumor progression, genetic alterations that were not detectable in the metastases were present in the primary tumor. These findings indicate that the primary tumor progresses genetically even after the metastasis has occurred, that is, the predominant clone of some primary tumor components no longer represents the metastatic clones that we investigated.
Using multiple microdissected specimens within individual cases that included primary and metastatic tumors, we have shown highly frequent LOH at 5q and 17p even in the lowest-grade portions (ie, BA lesions) and found identical alleles to be deleted in all portions examined in each affected primary tumor and metastatic tumor, even though quite divergent histopathologically. These results suggest that LOH at 5q and 17p may be crucial steps in the early phase of the development to lymph node metastasis and that they are retained throughout successive clonal evolutions. A similar phenomenon was previously reported by Boland et al23 based on an analysis of tumor progression schemes in colon cancers. They used multiple microdissected samples of colonic tumors showing cross-sections of the ‘adenoma–carcinoma sequence’ and detected a clear, abrupt occurrence of LOH at 5q at the transition phase from normal epithelium to adenoma. We observed LOH on at least one of the two loci on 5q and/or 17p uniformly in almost all cases examined (11/12; 92%). However, it is uncertain why such presumably early lesions are not present in all tumors. A number of cell types are thought to be potential precursors of lung cancers, and different initiation events may be involved depending on the differentiation pathway to which they are committed. Thus, certain cell types may not require inactivation of all putative tumor suppressor genes on 5q and 17p.
Previous studies have shown allelic loss at the 17p loci to be involved at a relatively early stage of NSCLC,8, 24, 25 and its loss may be associated with the genesis of NSCLC. p53 is believed to play a role as a ‘guardian’ that maintains the integrity of the genome by participating in the DNA damage checkpoints in the cell cycle. Inactivation of p53 has been reported to lead to increased frequency of mutations, chromosomal rearrangements, and abnormal chromosomal segregations.26, 27, 28, 29 Recent studies have suggested that the LOH at specific chromosomal loci, 1p, 3p, 5q, 9p, 17q, and 22q, is associated with a worse prognosis of NSCLC, although studies of patients from different populations have yielded conflicting results.14, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39 LOH at the APC/MCC gene cluster at chromosome 5q has been reported to correlate with poorer survival of patients with NSCLC.38 In the present study, the tumors were small but were associated with lymph node metastasis, and a worse prognosis was assumed. Seven of nine informative cases (78%) showed allelic losses at 5q in all foci, including BA lesions. Our previous study also concluded that this deletion is a relatively early event in the progression of adenocarcinoma of the lung.6 The high prevalence of 5q deletion in this study might indicate that 5q loss plays an important role in the progression of metastatic tumors and that it was determined in the early stage.
Although we showed frequent LOH at 18q in the tumors examined, approximately 40% of allelic losses were found in either metastatic lesions or invasive and metastatic lesions, not in all portions of each affected primary tumor and metastatic tumor examined. Therefore, LOH at 18q appears to have a role as late event in the metastatic progression of adenocarcinomas mixed BA and other subtypes of the lung. Shiseki et al8 reported that loss at 18q plays an important role in the progression of NSCLC based on a comparison of stage I NSCLC and brain metastases. Lymph node metastases, most malignant portions, were shown to carry 18q deletions at even higher frequency than 5q or 17p deletions in the present study. However, it should be noted that accumulation of LOH at 18q occurred at various stages of tumor progression within individual tumors toward lymph node metastasis, that is, some LOH at 18q occurred in BA lesions, and some in metastatic lesions. This indicated a clear distinction from LOH at 5q and 17p, especially at 5q. Since approximately 60% of 18q deletions are present in all portions of individual tumors, it remains unclear whether LOH at 18q has a role both as an early event and a late event or acts at various steps in tumors that progress to lymph node metastasis.
In summary, we examined the topographical distribution of LOH on 10 chromosome arms, and the results suggest that tumor cells accumulate genetic alterations as they evolve from the BA lesions to the invasive and metastatic lesions. Early occurrence of 5q and/or 17p deletions and successive clonal expansion during the progression of individual tumors was inferred. By contrast, LOH at 18q seemed to be acquired at various stages during tumor progression to metastasis. Similar studies analyzing more genetic loci in a larger number of cases are warranted. Furthermore, since the lung cancers resulted from various genetic and epigenetic alterations, it would also be interesting to examine the topographical differences in other genetic or epigenetic changes, such as DNA methylation.
References
Fearon ER, Vogelstein B . A genetic model for colorectal tumorigenesis. Cell 1990;61:759–767.
Travis WD, Colby TV, Corrin B, et al. Histological Typing of Lung and Pleural Tumours, 3rd edn. Springer-Verlag: Heidelberg, 1999.
Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med 1988;319:525–532.
Cho KR, Vogelstein B . Genetic alterations in the adenoma–carcinoma sequence. Cancer 1992;70:1727–1731.
Baker SJ, Preisinger AC, Jessup JM, et al. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res 1990;50:7717–7722.
Aoyagi Y, Yokose T, Minami Y, et al. Accumulation of losses of heterozygosity and multistep carcinogenesis in pulmonary adenocarcinoma. Cancer Res 2001;61:7950–7954.
Noguchi M, Morikawa A, Kawasaki M, et al. Small adenocarcinoma of the lung. Histologic characteristics and prognosis. Cancer 1995;75:2844–2852.
Shiseki M, Kohno T, Adachi J, et al. Comparative allelotype of early and advanced stage non-small cell lung carcinomas. Genes Chromosomes Cancer 1996;17:71–77.
Wistuba, II, Behrens C, Milchgrub S, et al. Sequential molecular abnormalities are involved in the multistage development of squamous cell lung carcinoma. Oncogene 1999;18:643–650.
Fidler IJ, Hart IR . Biological diversity in metastatic neoplasms: origins and implications. Science 1982;217:998–1003.
Weiss L . Metastasis of cancer: a conceptual history from antiquity to the 1990s. Cancer Metastasis Rev 2000;19, I-XI 193–383.
Sato N, Tsunoda H, Nishida M, et al. Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: possible sequence progression from benign endometrial cyst to endometrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res 2000;60:7052–7056.
Ohta M, Inoue H, Cotticelli MG, et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint is abnormal in digestive tract cancers. Cell 1996;84:587–597.
Sanchez-Cespedes M, Rosell R, Pifarre A, et al. Microsatellite alterations at 5q21, 11p13, and 11p155 do not predict survival in non-small cell lung cancer. Clin Cancer Res 1997;3:1229–1235.
Tsutsumi M, Tsai YC, Gonzalgo ML, et al. Early acquisition of homozygous deletions of p16/p19 during squamous cell carcinogenesis and genetic mosaicism in bladder cancer. Oncogene 1998;17:3021–3027.
Okami K, Cairns P, Westra WH, et al. Detailed deletion mapping at chromosome 9p21 in non-small cell lung cancer by microsatellite analysis and fluorescence in situ hybridization. Int J Cancer 1997;74:588–592.
Ogawara K, Miyakawa A, Shiba M, et al. Allelic loss of chromosome 13q14.3 in human oral cancer: correlation with lymph node metastasis. Int J Cancer 1998;79:312–317.
Goto A, Kanda H, Ishikawa Y, et al. Association of loss of heterozygosity at the p53 locus with chemoresistance in osteosarcomas. Jpn J Cancer Res 1998;89:539–547.
Takei K, Kohno T, Hamada K, et al. A novel tumor suppressor locus on chromosome 18q involved in the development of human lung cancer. Cancer Res 1998;58:3700–3705.
Anami Y, Takeuchi T, Mase K, et al. Amplotyping of microdissected, methanol-fixed lung carcinoma by arbitrarily primed polymerase chain reaction. Int J Cancer 2000;89:19–25.
Langenbach N, Kroiss MM, Ruschoff J, Schlegel J, et al. Assessment of microsatellite instability and loss of heterozygosity in sporadic keratoacanthomas. Arch Dermatol Res 1999;291:1–5.
Takamochi K, Ogura T, Suzuki K, et al. Loss of heterozygosity on chromosomes 9q and 16p in atypical adenomatous hyperplasia concomitant with adenocarcinoma of the lung. Am J Pathol 2001;159:1941–1948.
Boland CR, Sato J, Appelman HD, et al. Microallelotyping defines the sequence and tempo of allelic losses at tumour suppressor gene loci during colorectal cancer progression. Nat Med 1995;1:902–909.
Yatabe Y, Konishi H, Mitsudomi T, et al. Topographical distributions of allelic loss in individual non-small-cell lung cancers. Am J Pathol 2000;157:985–993.
Sasatomi E, Finkelstein SD, Woods JD, et al. Comparison of accumulated allele loss between primary tumor and lymph node metastasis in stage II non-small cell lung carcinoma: implications for the timing of lymph node metastasis and prognostic value. Cancer Res 2002;62:2681–2689.
Havre PA, Yuan J, Hedrick L, et al. p53 inactivation by HPV16 E6 results in increased mutagenesis in human cells. Cancer Res 1995;55:4420–4424.
Fukasawa K, Choi T, Kuriyama R, et al. Abnormal centrosome amplification in the absence of p53. Science 1996;271:1744–1747.
Bertrand P, Rouillard D, Boulet A, et al. Increase of spontaneous intrachromosomal homologous recombination in mammalian cells expressing a mutant p53 protein. Oncogene 1997;14:1117–1122.
Kohno T, Yokota J . How many tumor suppressor genes are involved in human lung carcinogenesis? Carcinogenesis 1999;20:1403–1410.
Thiberville L, Bourguignon J, Metayer J, et al. Frequency and prognostic evaluation of 3p21–22 allelic losses in non-small-cell lung cancer. Int J Cancer 1995;64:371–377.
Sanz-Ortega J, Bryant B, Sanz-Esponera J, et al. LOH at the APC/MCC gene (5Q21) is frequent in early stages of non-small cell lung cancer. Pathol Res Pract 1999;195:677–680.
Tomizawa Y, Adachi J, Kohno T, et al. Prognostic significance of allelic imbalances on chromosome 9p in stage I non-small cell lung carcinoma. Clin Cancer Res 1999;5:1139–1146.
Fong KM, Kida Y, Zimmerman PV, et al. MYCL genotypes and loss of heterozygosity in non-small-cell lung cancer. Br J Cancer 1996;74:1975–1978.
Chizhikov V, Zborovskaya I, Laktionov K, et al. Two consistently deleted regions within chromosome 1p32-pter in human non-small cell lung cancer. Mol Carcinog 2001;30:151–158.
Mitsudomi T, Oyama T, Nishida K, et al. Loss of heterozygosity at 3p in non-small cell lung cancer and its prognostic implication. Clin Cancer Res 1996;2:1185–1189.
Osaki T, Oyama T, Inoue M, et al. Molecular biological markers and micrometastasis in resected non-small-cell lung cancer. Prognostic implications. Jpn J Thorac Cardiovasc Surg 2001;49:545–551.
Burke L, Khan MA, Freedman AN, et al. Allelic deletion analysis of the FHIT gene predicts poor survival in non-small cell lung cancer. Cancer Res 1998;58:2533–2536.
Fong KM, Zimmerman PV, Smith PJ . Tumor progression and loss of heterozygosity at 5q and 18q in non-small cell lung cancer. Cancer Res 1995;55:220–223.
Fong KM, Kida Y, Zimmerman PV, et al. Loss of heterozygosity frequently affects chromosome 17q in non-small cell lung cancer. Cancer Res 1995;55:4268–4272.
Acknowledgements
This work was supported by a Grant-in-Aid for Cancer Research from the Ministry for Health and Welfare 11–12 and by a Grant-in-Aid for Scientific Research Expenses for Health Labour and Welfare Programs and the Foundation for the Promotion of Cancer Research, and by the 2nd-Term Comprehensive 10-year Strategy for Cancer Control.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yoshikawal, T., Aoyagi, Y., Kodama, K. et al. Topographical distribution of allelic loss in individual lung adenocarcinomas with lymph node metastases. Mod Pathol 17, 204–213 (2004). https://doi.org/10.1038/modpathol.3800035
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.3800035
