
Abstract
Angiotensin II (AngII) plays an important role in renal damage by acting on hemodynamics, cell-growth, proliferation, and fibrosis, mainly by effects on the AngII type 1 (AT1) receptor. The AT1 receptor activates several intracellular signaling molecules such as mitogen-activated protein kinases extracellular signal-regulated kinase (ERK) and p38, but their role in AngII-mediated renal damage is not well characterized. We therefore investigated whether pharmacologic blockade of ERK and p38 could prevent renal damage in high-renin homozygous transgenic rats (Ren2), with the effects of an AT1 receptor antagonist (AT1-RA) as a reference. Seven-week-old homozygous Ren2 rats were treated with low-dose AT1-RA candesartan, ERK inhibitor tyrphostin, or p38 inhibitor SB239063 for 4 weeks. Untreated Ren2 and sd rats served as controls. Blood pressure was measured at 7 and 11 weeks. At 11 weeks, plasma renin activity (PRA) and serum aldosterone were determined, and the animals were killed. Kidney sections were scored for glomerular and interstitial smooth muscle actin and glomerular desmin expression as early markers for renal damage. Mesangial matrix expansion was determined as a marker for structural damage. PRA and aldosterone levels were elevated in untreated Ren2 rats in comparison to sd controls. AT1-RA further increased PRA but decreased aldosterone. All parameters of renal damage were elevated in untreated Ren2 rats. Blood pressure was not elevated at week 7 in Ren2 and not affected by either treatment. Mild signs of hypertensive damage were found in untreated Ren2 rats. All interventions significantly diminished damage to glomerular epithelium and interstitium. In addition, AT1 receptor and p38 blockade reduced mesangial matrix expansion. In homozygous Ren2 rats, renal damage was ameliorated by a nonhypotensive dose of an AT1-RA and, similarly, by blockade of ERK or p38. This suggests that ERK and p38 are involved in AngII-mediated renal damage.
Similar content being viewed by others

Main
The renin-angiotensin system (RAS) plays a crucial role in renal pathophysiology. Angiotensin II (AngII), a main effector molecule of the RAS, exerts vasoactive effects but also functions as a growth factor that regulates cell proliferation, hypertrophy, and extracellular matrix production. Presumably, the latter processes are involved in the development of (pre-)fibrotic glomerular and interstitial lesions in various renal disorders (Anderson et al, 1993; Gomez-Garre et al, 1996; Wolf et al, 1992).
Several of the deleterious effects of AngII act via the AngII type 1 (AT1) receptor and can be reversed by AT1 receptor blockade (Border et al, 1998; Mezzano et al, 2001; Wu et al, 1997). Downstream of the AT1 receptor, the mitogen-activated protein (MAP)-kinase superfamily of intracellular signaling molecules is an important pathway. Two relevant members of this family are extracellular signal-regulated kinase (ERK) and p38 MAP-kinase. AngII activates ERK via the AT1 receptor in mesangial cells, glomerular endothelial cells, and in proximal tubulus cells (Huwiler et al, 1995; Terada et al, 1994; Wolf et al, 1996). Activation of ERK is considered to play a key role in mitogenesis and hypertrophy (Treisman, 1996). AngII activates p38 in various cell types, among others in vascular smooth muscle cells (Ingram et al, 2000; Ushio-Fukai et al, 1998). Activation of p38 by AngII is important in vascular hypertrophy, but also in transforming growth factor (TGF)β-mediated signaling and extracellular matrix induction in glomerular mesangial cells (Choi, 2000; Ushio-Fukai et al, 1998). Furthermore, in diabetic rats, glomerular activation of p38 was increased compared with nondiabetic and insulin-treated diabetic rats (Dunlop et al, 2000). However, the functional significance of ERK and p38 activation in AngII-mediated renal damage has remained unclear.
We therefore investigated the effects of selective blockade of ERK and p38 in a high-renin rat model of renal damage, with rats treated with the AT1 receptor antagonist (AT1-RA) candesartan as a positive control for protection against renal damage. Because we were specifically interested in the intrarenal effects of RAS activation (as opposed to the renal effects of hypertension), we used homozygous TGR(mRen2)27 rats. Unlike heterozygous rats in which malignant hypertension develops with the corresponding renal abnormalities, in homozygous Ren2 rats, after an initial period of hypertension at an early age, blood pressure returns to normal values, secondary to cardiac dysfunction. To also avoid the confounding effects of blood pressure reduction in the candesartan controls, a nonhypotensive dose was used.
Results
Model Characteristics
In homozygous TGR(mRen2)27 rats, systolic blood pressure (SBP) was elevated at 6 and 10 weeks, and decreased at 13 weeks, compared with sd controls (Fig. 1). Urine protein to creatinine ratios were higher in Ren2 rats than in sd rats at 7 weeks (0.19 ± 0.04 versus 0.13 ± 0.02; mean ± sem), 8 weeks (0.48 ± 0.13 versus 0.09 ± 0.01), and 11 weeks (0.22 ± 0.09 versus 0.13 ± 0.02); however, the difference reached statistical significance at week 8 only (p < 0.01).

Time course of systolic blood pressure in homozygous Ren2 and sd rats (mean ± sem; *p < 0.05). Open circles indicate Ren2 rats; solid circles indicate sd rats.
Clinical Parameters
As shown in Table 1, in untreated homozygous Ren2 rats, plasma renin activity (PRA) and aldosterone levels were increased. There was no significant difference in SBP between untreated Ren2 and sd rats at 7 or 11 weeks (SBP before treatment and SBP at sacrifice, respectively). As expected, the AT1-RA candesartan further increased PRA and decreased aldosterone levels in Ren2 rats. The fact that the administered dose of candesartan was nonhypotensive was confirmed using a paired samples t test (p = not significant). Renal function of Ren2 rats treated with candesartan was slightly reduced compared with untreated Ren2 rats, as indicated by plasma creatinine (6.54 [6.19 to 8.64] versus 5.60 [4.20 to 6.19] mg/L; p = 0.05).
ERK and p38 inhibition did not affect PRA, aldosterone, or creatinine levels, or SBP. Cholesterol levels in untreated Ren2 rats were equal to sd controls and in any treated Ren2 group compared with untreated. N-terminal atrial natriuretic peptide (N-ANP), a marker for heart failure, was increased in untreated Ren2 compared with sd controls. Treatment with candesartan or the ERK inhibitor reduced N-ANP levels, the p38-inhibitor–treated rats showed only a trend but no significant effect. Body weight of the Ren2 rats was significantly decreased compared with sd controls; candesartan-treated Ren2 rats showed increased body weight compared with untreated Ren2. Treatment with ERK or p38 inhibitor had no effect on body weight. In the untreated Ren2 group, one animal died; two animals died in the ERK-inhibitor–treated group and one in the group treated with the p38 inhibitor.
Morphology, Immunohistochemistry, and PCR
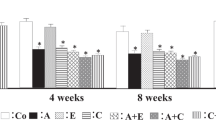
In the untreated Ren2 rats, no renal vascular damage was found, and, morphologically, the renal interstitium was largely intact. Glomerular changes were especially found in the cortico-medullary regions and consisted of mild but typical signs of hypertensive glomerulopathy, including thickening of the capillary walls and thickening of the mesangium. The latter was predominant in the glomerular vascular pole. Adhesion of the visceral epithelial cells to Bowman’s capsule was occasionally present. In the more advanced lesions, segmental scarring of the glomerular tuft was observed. Renal damage, as determined by scoring early renal phenotypic changes (α–smooth muscle actin [SMA] and desmin) and structural damage (mesangial matrix expansion) were significantly increased in untreated Ren2 rats compared with untreated sd rats. Glomerular α-SMA expression was not altered by either the ERK or p38 inhibitor or candesartan (p = not significant, data not shown). In the interstitium, on the other hand, α-SMA expression decreased in the ERK-, p38- and AT1-RA–treated groups, indicating reduced myofibroblast transformation (Fig. 2). Furthermore, both AT1 receptor and MAP-kinase inhibition resulted in decreased glomerular desmin expression (Fig. 3). Mesangial matrix expansion was significantly reduced by candesartan and SB239063 but not by the ERK inhibitor (Fig. 4).

Immunostaining and morphometric analysis for α-smooth muscle actin (SMA) in the renal interstitium. SMA score was higher in untreated Ren2 rats than in untreated sd rats, and significantly lower in Ren2 rats receiving an angiotensin II type 1 receptor antagonist (AT1-RA), extracellular signal-regulated kinase (ERK) inhibitor, or p38 inhibitor, when compared with untreated Ren2 rats (diagram). Figures represent immunohistochemistry for SMA in the interstitium of untreated sd rats (A), and Ren2 rats (B: untreated, C: AT1-RA, D: ERK inhibitor). Results for the group that received a p38 inhibitor were comparable to D. Original magnification: × 200. *p < 0.05 vs untreated sd rats. **p < 0.05 vs untreated Ren2 rats.

Desmin expression, indicating damage to the glomerular epithelium (podocytes), was increased in untreated Ren2 rats. Treatment with an angiotensin II type 1 receptor antagonist (AT1-RA), as well as an extracellular signal-regulated kinase (ERK) or p38 inhibitor significantly reduced desmin scores. Figures represent glomeruli of sd (A) and Ren2 rats, either untreated (B), or treated with an AT1-RA (C) or p38 inhibitor (D). Results for the group that was treated with an ERK inhibitor were comparable to D. Original magnification: × 350. *p < 0.05 vs sd. **p < 0.05 vs Ren2.

Mesangial matrix expansion (MME) was increased in the untreated Ren2 group. The angiotensin II type 1 receptor antagonist (AT1-RA) and p38 inhibitor, but not the extracellular signal-regulated kinase (ERK) inhibitor, significantly reduced MME. Representative images after periodic acid-Schiff staining, demonstrating the extent of MME. (A) Untreated sd rats, (B) Untreated Ren2 rats, (C) Ren2 rats treated with AT1-RA, (D) Ren2 rats treated with p38 inhibitor. Original magnification: × 350. *p < 0.05 vs sd. **p < 0.05 vs Ren2.
Immunohistochemistry for renin revealed subtle de novo renin expression in some tubular epithelial cells in untreated Ren2 rats. In the other groups, tubuli were negative. Ren2 rats showed decreased renin expression in the afferent arteriolar wall compared with sd controls. Treatment with candesartan resulted in increased renin expression in the juxtaglomerular apparatus, both in Ren2 and in sd animals. Treatment with the ERK or p38 inhibitor had no effect on renin expression in the arteriolar wall (data not shown). These findings are in line with previous data (Lee et al, 1995). PCR for TGF-β1 revealed no difference between Ren2 and sd (66.3 ± 17.4 versus 61.7 ± 15.5% versus glyceraldehyde-3-phosphate dehydrogenase [GAPDH]), nor between treated and untreated Ren2 groups (candesartan: 55.2 ± 14.8; ERK inhibition: 82.4 ± 8.8; and p38 inhibitor: 75.3 ± 5.6% versus GAPDH).
Discussion
The major finding of this study is the significant amelioration of early prefibrotic changes and structural renal damage by specific blockade of MAP-kinases ERK and p38, similar to the effects of the AT1-RA candesartan. This suggests that ERK and p38 are involved in AngII-mediated pathways of renal damage, mediated by intrarenal activity of the RAS.
In this study we used rats overexpressing the Ren2 gene, and accordingly in untreated Ren2 rats, plasma renin activity was significantly increased, confirming the genetically enhanced activity of the RAS in these rats. It has been shown that circulating renin can be taken up by tissues, leading to local AngII formation, which might be involved in the prefibrotic transdifferentiation of renal tubules (Langheinrich et al, 1996; Muller et al, 1998). Moreover, using immunohistochemistry we found some local tubular renin expression in the kidneys of untreated Ren2 rats, while tubuli of untreated sd and all treated Ren2 rats were negative. This raises the possibility that increased local renin is also relevant to enhanced generation of AngII, which would be in line with data by Springate et al (1997). Our data, albeit only subtle local renal renin expression, can be considered in line with prior observations in this model (Bader et al, 1992; Langheinrich et al, 1996).
Before intervention, we measured SBP and urinary protein in a time-course characterization of the homozygous TGR(mRen2)27 model. The time course of SBP in Ren2 rats demonstrates a period of high blood pressure at young age, followed by a decreasing blood pressure. The increased N-ANP levels in our study confirm prior studies by our group suggesting that the poor cardiac function of homozygous as opposed to heterozygous Ren2 rats precludes the development of malignant hypertension (De Boer et al, 2001; Lee et al, 1996). In contrast to heterozygous rats, in our homozygous Ren2 rats, proteinuria if anything was mild. This is presumably due to the early decline in blood pressure to low normotensive values, secondary to early development of heart failure, as blood pressure has been shown to be an important determinant of renal damage in the Ren2 model (Langheinrich et al, 1996; Witte et al, 1999). However, data on proteinuria in our study are not complete and would need confirmation from further studies.
Morphologically, the severity of renal damage in these young-adult homozygous Ren2 rats was relatively mild and consisted of prefibrotic changes in the glomeruli as well as the interstitium, in addition to damaged glomeruli in the cortico-medullary areas, which was made apparent by the increased expression of desmin and α-SMA. Prior studies demonstrating increased expression of desmin and α-SMA after infusion of exogenous angiotensin indicate that this is compatible with AngII-induced renal damage (Johnson et al, 1992a; 1992b).
The renal phenotype observed in the present study is somewhat at variance with previous studies in heterozygous Ren2 rats in which renal damage displays all characteristics of malignant hypertension in accord with the clinical picture in these animals (Lee et al, 1996). Our current data indicate that in the absence of hypertension, AngII-mediated damage is mild (at least within the time frame of this study), with relatively mild renal abnormalities despite gross elevation of PRA.
A nonhypotensive dose of the AT1-RA candesartan was used as a positive control for the protection against RAS-mediated renal damage. A nonhypotensive dose was chosen to be able to dissociate the effects of AT1 blockade as such from those of blood pressure reduction (Springate et al, 1997). Via the AT1 receptor, AngII activates the transcription factor activator protein-1, which is responsible for fibrosis and proliferation by the formation of MCP-1, fibronectin, TGF-β, and c-Fos (Ruiz-Ortega et al, 2001). Activation of activator protein-1 is mediated by protein kinase C and protein tyrosine kinase, as well as MAP-kinases ERK, c-JUN NH2-terminal kinase, and p38 MAP-kinase (Matsubara, 1998; Tharaux et al, 2000; Viedt et al, 2000). As the MAP-kinases ERK and p38 act downstream of the AT1-receptor, we consider candesartan a positive control for this experiment. The similarity of the findings in the groups treated with MAP-kinase inhibition and candesartan, respectively, supports the assumption that the renal profibrotic changes in this model are mainly mediated by AngII via the AT1 receptor and subsequent MAP-kinase activation. This is in accordance with earlier studies that showed that AngII can induce tissue damage independent of blood pressure, a process that is mediated by the AT1 receptor (Border and Noble, 1998; Mezzano et al, 2001; Wu et al, 1997). However, MAP-kinases are involved in G-protein–dependent pathways that are activated by other neurohormones as well, such as endothelin-I and norepinephrine; therefore, we cannot completely exclude the possibility that the renoprotective effects of MAP-kinase inhibition in our study are (partly) mediated by interference in non-AngII pathways.
Only a single dose of each MAP-kinase inhibitor was used in the present study, so information on dose-response is lacking. Although we have no marker of renal inhibition of ERK or p38, we found a significant renoprotective effect, similar to that of candesartan (ie, our positive control), which demonstrates the pharmacologic efficacy of the regimens.
How could MAP-kinase inhibitors protect against the development of renal fibrosis? In vitro studies in human mesangial cells showed that selective inhibition of p38 completely abolishes stretch-induced fibronectin and TGF-β1 production (Gruden et al, 2000) and that ERK plays a role in TGF-β1–stimulated collagen I expression (Hayashida et al, 1999) and in connective tissue growth factor induction by TGF-β (Chen et al, 2002). In our experiment, PCR for TGF-β1 showed no difference between Ren2 and sd, nor between treated and untreated Ren2 groups. The fact that TGF-β expression was not increased in Ren2 compared with sd animals could be explained by the fact that these animals show only mild prefibrotic renal damage. In the hearts of Ren2 rats, increased TGF-β mRNA is described by Pinto et al (2000).
Moreover, both ERK and p38 play a role in various in vivo models of renal disease, eg, anti-Thy1 glomerulonephritis (Bokemeyer et al, 2000) and salt-loaded stroke-prone spontaneously hypertensive rats. In the latter model, selective p38 MAPK inhibition by SB239063 reduced hypertensive end-organ (ie, renal) damage and premature mortality. In accord with our present findings, renal protective effects were observed in the absence of significant effects on blood pressure (Behr et al, 2001).
Interestingly, blockade of ERK or p38, in contrast to AT1 receptor blockade, had no effect on PRA or plasma aldosterone levels. This suggests that the feedback mechanism regulating the RAS is mediated by the AT1 receptor but not by the downstream molecules ERK or p38, at least not at the doses used in the current study. Although our study was not set up to test this hypothesis, these findings appear in contrast with a previous study demonstrating p38 MAP-kinase to be involved in renin production mediated by cyclo-oxygenase 2 (Cheng et al, 2000).
We used an epidermal growth factor receptor (EGF-R) inhibitor (tyrphostin A46) to inhibit ERK activation. Although EGF-R is directly upstream of ERK in the MAP-kinase cascade, the effects of tyrphostin A46 might be ascribed to the inhibition of EGF-R. However, in various cell types (eg, fibroblasts and epithelial cells), ERK activation is directly modulated by inhibition and activation of EGF-R (Murasawa et al, 1998; Pai et al, 1998).
In conclusion, inhibition of the MAP-kinases ERK and p38 decreased renal damage in the homozygous Ren2 model, comparable to the AT1-RA candesartan, in a non–blood pressure–dependent fashion. These data suggest that ERK and p38 are involved in AngII-mediated fibrotic renal damage. Further studies are needed to better characterize the role of these intracellular signaling pathways in the development of RAS-mediated renal damage.
Materials and Methods
Model Characterization
To study the time course of SBP and proteinuria in the homozygous Ren2 model, we performed two preliminary experiments in male homozygous TGR(mRen2)27 rats (n = 5 per group), using Sprague Dawley Hannover (n = 7 per group) rats as controls. We measured SBP by tail-cuff method under 2% to 4% isoflurane anesthesia, at three different time points (6, 10, and 13 weeks). Also, we determined urine albumin/creatinine ratio at Weeks 7, 8, and 13 in a urine sample drawn before death, as a measure for urinary protein excretion. This ratio was used because high stress-induced mortality in homozygous Ren2 rats precludes frequent use of metabolic cages. Urinary protein levels were measured using the pyrogallol red molybdate method. Urine creatinine levels were determined colorimetrically (Sigma Chemical Company, St Louis, Missouri).
Study Design
Experiments were conducted in 7-week-old male homozygous TGR(mRen2)27 rats. Age-matched Sprague-Dawley (sd) rats served as controls. Homozygous Ren2 rats and sd rats were purchased from the Max Delbrück Center for Molecular Medicine, Berlin-Buch, Berlin, Germany. Transgenic status was confirmed by Southern Blotting. All animals were housed in a light- and temperature-controlled environment and were fed standard rat chow with free access to tap water. All procedures were approved by the committee for animal experiments of the University of Groningen.
At 7 weeks, Ren2 animals were divided into four groups (n = 7), which were either left untreated or treated for 4 weeks with a low dose of the AT1-RA candesartan, the EGF-R blocker tyrphostin as an inhibitor of ERK or the p38 MAPK inhibitor SB 239063 (see below). Untreated sd rats served as negative controls (n = 7).
The animals were killed at 11 weeks. PRA and aldosterone were determined (see below), and creatinine and cholesterol levels were measured using ELISA. Kidneys were weighed and embedded in paraffin for immunohistochemical analysis. For technical reasons (low blood pressure and oligo-anuria during the termination session), no urine could be obtained for protein/creatinine ratio in these animals.
Drugs and Minipumps
The AT1-RA candesartan (a gift from AstraZeneca, Zoetermeer, the Netherlands) was dissolved in 0.1 M NaHCO3 in saline (0.9% NaCl), according to the manufacturer’s recommendations. The final dose administered was 0.05 mg/kg/day. Furthermore, we used the EGF-R blocker tyrphostin A46 (Sanvertech, Heerhugowaard, the Netherlands), a known inhibitor of ERK activity, which was dissolved in 0.01% dimethyl sulfoxide and H2O, according to manufacturer’s guidelines (De Boer et al, 2001; Murasawa et al, 1998). Final dose administered was 600 μg/kg/day. The p38 MAPK inhibitor SB239063 (trans-1-[4-hydroxycyclohexyl]-4-[4-fluorophenyl]-5-[2-methoxypyridimidin-4-yl]imidazole; IC50 = 44 nm versus p38α), kindly provided by Dr. R.N. Willette (Department of Cardiovascular Pharmacology, GlaxoSmithKline, King of Prussia, Pennsylvania), was dissolved in 0.1% dimethyl sulfoxide in H2O, with 50% polyethylene glycol (PEG) 400 (Underwood et al, 2000). Final dose administered was 15 mg/kg/day. All drugs were administered by osmotic minipumps (types 2004 and 2ML4, depending on dose regimen; Alzet, Palo Alto, California). Minipumps were aseptically filled by injection of the fluid containing the drug, and weighed before and after filling. The volume weight injected was calculated to control for adequate filling of the pumps. Rats were anesthetized with 2% to 4% isoflurane in a gas mixture of N2O/O2 (2/1). Candesartan- and SB239063-filled minipumps were inserted subcutaneously, and minipumps filled with tyrphostin A46 were placed intraperitoneally. At killing, the minipumps were collected and weighed to determine the volume that was dispensed.
Renin, Aldosterone, and Plasma N-ANP Levels
Blood was obtained from the abdominal aorta, anticoagulated with EDTA, and immediately centrifuged (4° C, 1500 × g for 15 minutes). Plasma was stored at −80° C until assayed. PRA was measured by determining the amount of AngI generated from angiotensinogen with radioimmunoassay.
For measurement of plasma aldosterone concentration (normal: 50 to 250 pg/ml), a commercially available kit (Coat-a-Count, Diagnostic Products Corporation, Los Angeles, California) was used.
Plasma N-ANP was measured with a commercially available radioimmunoassay kit (Bioptop, Oulu, Finland). All assays were previously described (van Veldhuisen et al, 1998).
Immunohistochemistry
Four-μm paraffin sections were dewaxed and washed in PBS. Heat-induced antigen retrieval was achieved by incubation in 0.1 M Tris/HCl buffer (pH = 9.0) overnight at 80° C. For immunohistochemistry, a DAKO Autostainer was used (DAKO Corporation, Carpinteria, California). Sections were washed in PBS and treated with endogenous peroxidase blocking reagent containing 0.03% H2O2/sodium azide for 5 minutes. Slides were then incubated with primary antibodies against α-SMA (clone 1A4, Sigma Chemical Company, St. Louis, Missouri), renin (a kind gift from Professor T. Inagami, Vanderbilt University School of Medicine, Nashville, Tennessee) or desmin (Dakopatts, DAKO, Glostrup, Denmark), diluted in PBS containing 1% bovine serum albumin (BSA) for 60 minutes at room temperature. Binding was detected by sequential incubation with peroxidase-labeled rabbit antimouse, goat antirabbit, and rabbit antigoat polyclonals (Dakopatts, DAKO), in the presence of 1% normal rat serum for 30 minutes. The peroxidase activity was visualized using 3,3′-diaminobenzidine tetrahydrochloride (DAB+, category no. K3468; DAKO) for 10 minutes. Sections stained for desmin were counterstained with hematoxylin for 2 seconds, made blue in running tap water, and mounted with Kaiser’s glycerin gelatin.
PCR for TGF-β1
Total RNA from cell pellets or frozen tissue containing > 1.106 cells was isolated with TRIZOL using manufacturer’s instructions. RNA was reverse transcribed according to the manufacturer’s instructions in 50 μl using 1 mm of each dNTP, 25 μg/ml oligo dT24 primer, 10 U/μl M-MLV reverse transcriptase (Invitrogen, Carlsbad, California), and the reaction buffer supplied by the manufacturer. PCR was performed in a 30-μl reaction with 1 unit of TaqDNA polymerase (Amersham Biosciences, Buckinghamshire, United Kingdom), the reaction buffer provided by the manufacturer and 1 μl cDNA. PCR cycles consisted of 30 seconds 94° C, 45 seconds 55° C, and 45 seconds 72° C. The final extension step consisted of 7 minutes at 72° C. PCR samples were analyzed on a 1.2% agarose gel after an increasing number of PCR cycles. Primer sequences used were as follows: TGF-β1 F 5′-AGAAGAACTGCTGTGTACGG-3′, R 5′-ACCCACGT-AGTAGACGATGG-3′, GAPDH F 5′-ACTCAGAAGACTGTGGATGG-3′, R 5′-GTTGCTGTTGAAGTCACAGG-3′. GAPDH was used as an RNA loading control. In each experiment, positive and negative controls were included. Images were prepared and analyzed using Geldoc software (Quantity One, Bio-Rad, Veenendaal, the Netherlands).
Morphological Measurements
The extent of glomerular α-SMA expression was measured using computerized morphometry. For that purpose, 30 glomeruli per section were screened using a light microscope equipped with a camera device connected to a computer system. The image of a given glomerulus present on the screen was traced with a cursor along Bowman’s capsule. Subsequently, the total surface with brown precipitate was measured and divided by the total surface of the glomerulus. Per cortical section the average score of all glomeruli was calculated.
The extent of interstitial α-SMA expression was also determined by morphometry. Thirty rectangular fields without any glomeruli or arteries were selected, and again the amount of total brown precipitate was measured and represented as a percentage of the total selected area. The ultimate interstitial α-SMA score was calculated by the average of all fields per section. All measurements were performed by a blinded observer (MHdB).
Desmin expression was scored semi-quantitatively by a blinded observer (LMV). This was achieved by estimating the percentage of desmin-positive epithelial cells located in the outer cell layer of the glomerular tuft. Semi-quantitative staining scores depended on the percentage of the glomerular edge showing positive staining: 0, 0% to 5% stained; 1, 5% to 25% stained; 2, 25% to 50%; 3, 50% to 75%; 4, > 75% (Joles et al, 2000). Thirty glomeruli were scored, and final desmin score was acquired by calculating the mean score.
Analysis of Structural Changes
Periodic acid-Schiff–stained sections were evaluated for morphologic changes, ie, the extent of mesangial matrix expansion. Sections were scored semi-quantitatively on a scale of 0 to 4+. If 25% of the glomerulus was affected, a score of 1+ was adjudged, 50% was scored as 2+, 75% as 3+, and 100% as 4+. A total of 50 glomeruli per animal was scored by a blinded observer (HvG), moving from cortex to medulla and vice versa. The ultimate score per animal was obtained by multiplying the degree of change by the percentage of glomeruli with the same degree of injury and addition of these scores.
Statistical Analysis
Data are presented as median and 95% confidence interval of the median. Differences between the untreated Ren2 and sd groups were analyzed using t tests with independent samples or Mann-Whitney U tests. Results of the various treatments were compared with the untreated Ren2 group using ANOVA with two-sided Dunnett post hoc analysis. A value of p < 0.05 was considered statistically significant. Data were analyzed using SPSS 10.0 statistical software package (SPSS Inc., Chicago, Illinois).
References
Anderson PW, Do YS, and Hsueh WA (1993). Angiotensin II causes mesangial cell hypertrophy. Hypertension 21: 29–35.
Bader M, Zhao Y, Sander M, Lee MA, Bachmann J, Bohm M, Djavidani B, Peters J, Mullins JJ, and Ganten D (1992). Role of tissue renin in the pathophysiology of hypertension in TGR(mREN2)27 rats. Hypertension 19: 681–686.
Behr TM, Nerurkar SS, Nelson AH, Coatney RW, Woods TN, Sulpizio A, Chandra S, Brooks DP, Kumar S, Lee JC, Ohlstein EH, Angermann CE, Adams JL, Sisko J, Sackner-Bernstein JD, and Willette RN (2001). Hypertensive end-organ damage and premature mortality are p38 mitogen-activated protein kinase-dependent in a rat model of cardiac hypertrophy and dysfunction. Circulation 104: 1292–1298.
Bokemeyer D, Ostendorf T, Kunter U, Lindemann M, Kramer HJ, and Floege J (2000). Differential activation of mitogen-activated protein kinases in experimental mesangioproliferative glomerulonephritis. J Am Soc Nephrol 11: 232–240.
Border WA and Noble NA (1998). Interactions of transforming growth factor-beta and angiotensin II in renal fibrosis. Hypertension 31: 181–188.
Chen Y, Blom IE, Sa S, Goldschmeding R, Abraham DJ, and Leask A (2002). CTGF expression in mesangial cells: Involvement of SMADs, MAP kinase, and PKC. Kidney Int 62: 1149–1159.
Cheng HF, Wang JL, Zhang MZ, McKanna JA, and Harris RC (2000). Role of p38 in the regulation of renal cortical cyclooxygenase-2 expression by extracellular chloride. J Clin Invest 106: 681–688.
Choi ME (2000). Mechanism of transforming growth factor-beta1 signaling. Kidney Int 77 (Suppl): 53–58.
De Boer RA, Pokharel S, Flesch M, Suurmeijer AJH, Van Kampen DA, Boomsma F, van Gilst W, van Veldhuisen DJ, and Pinto YM (2001). Inhibition of extracellular signal-reagulated kinase (ERK) prevents progressive LV remodeling in hypertensive heart failure (Abstract). Eur Heart J 22: 680.
Dunlop ME and Muggli EE (2000). Small heat shock protein alteration provides a mechanism to reduce mesangial cell contractility in diabetes and oxidative stress. Kidney Int 57: 464–475.
Gomez-Garre D, Ruiz-Ortega M, Ortego M, Largo R, Lopez-Armada MJ, Plaza JJ, Gonzalez E, and Egido J (1996). Effects and interactions of endothelin-1 and angiotensin II on matrix protein expression and synthesis and mesangial cell growth. Hypertension 27: 885–892.
Gruden G, Zonca S, Hayward A, Thomas S, Maestrini S, Gnudi L, and Viberti GC (2000). Mechanical stretch-induced fibronectin and transforming growth factor-beta1 production in human mesangial cells is p38 mitogen-activated protein kinase-dependent. Diabetes 49: 655–661.
Hayashida T, Poncelet AC, Hubchak SC, and Schnaper HW (1999). TGF-beta1 activates MAP kinase in human mesangial cells: A possible role in collagen expression. Kidney Int 56: 1710–1720.
Huwiler A, Stabel S, Fabbro D, and Pfeilschifter J (1995). Platelet-derived growth factor and angiotensin II stimulate the mitogen-activated protein kinase cascade in renal mesangial cells: Comparison of hypertrophic and hyperplastic agonists. Biochem J 305: 777–784.
Ingram AJ, James L, Ly H, Thai K, Cai L, and Scholey JW (2000). Nitric oxide modulates stretch activation of mitogen-activated protein kinases in mesangial cells. Kidney Int 58: 1067–1077.
Johnson RJ, Alpers CE, Yoshimura A, Lombardi D, Pritzl P, Floege J, and Schwartz SM (1992a). Renal injury from angiotensin II-mediated hypertension. Hypertension 19: 464–474.
Johnson RJ, Floege J, Yoshimura A, Iida H, Couser WG, and Alpers CE (1992b). The activated mesangial cell: A glomerular “myofibroblast”? J Am Soc Nephrol 2: S190–S197.
Joles JA, Kunter U, Janssen U, Kriz W, Rabelink TJ, Koomans HA, and Floege J (2000). Early mechanisms of renal injury in hypercholesterolemic or hypertriglyceridemic rats. J Am Soc Nephrol 11: 669–683.
Langheinrich M, Lee MA, Bohm M, Pinto YM, Ganten D, and Paul M (1996). The hypertensive Ren-2 transgenic rat TGR (mREN2)27 in hypertension research. Characteristics and functional aspects. Am J Hypertens 9: 506–512.
Lee MA, Bohm M, Kim S, Bachmann S, Bachmann J, Bader M, and Ganten D (1995). Differential gene expression of renin and angiotensinogen in the TGR(mREN-2)27 transgenic rat. Hypertension 25: 570–580.
Lee MA, Bohm M, Paul M, Bader M, Ganten U, and Ganten D (1996). Physiological characterization of the hypertensive transgenic rat TGR(mREN2)27. Am J Physiol 270: E919–E929.
Matsubara H (1998). Pathophysiological role of angiotensin II type 2 receptor in cardiovascular and renal diseases. Circ Res 83: 1182–1191.
Mezzano SA, Ruiz-Ortega M, and Egido J (2001). Angiotensin II and renal fibrosis. Hypertension 38: 635–638.
Muller DN, Fischli W, Clozel JP, Hilgers KF, Bohlender J, Menard J, Busjahn A, Ganten D, and Luft FC (1998). Local angiotensin II generation in the rat heart: Role of renin uptake. Circ Res 82: 13–20.
Murasawa S, Mori Y, Nozawa Y, Gotoh N, Shibuya M, Masaki H, Maruyama K, Tsutsumi Y, Moriguchi Y, Shibazaki Y, Tanaka Y, Iwasaka T, Inada M, and Matsubara H (1998). Angiotensin II type 1 receptor-induced extracellular signal-regulated protein kinase activation is mediated by Ca2+/calmodulin-dependent transactivation of epidermal growth factor receptor. Circ Res 82: 1338–1348.
Pai R, Ohta M, Itani RM, Sarfeh IJ, and Tarnawski AS (1998). Induction of mitogen-activated protein kinase signal transduction pathway during gastric ulcer healing in rats. Gastroenterology 114: 706–713.
Pinto YM, Pinto-Sietsma SJ, Philipp T, Engler S, Kossamehl P, Hocher B, Marquardt H, Sethmann S, Lauster R, Merker HJ, and Paul M (2000). Reduction in left ventricular messenger RNA for transforming growth factor beta(1) attenuates left ventricular fibrosis and improves survival without lowering blood pressure in the hypertensive TGR(mRen2)27 rat. Hypertension 36: 747–754.
Ruiz-Ortega M, Lorenzo O, Suzuki Y, Ruperez M, and Egido J (2001). Proinflammatory actions of angiotensins. Curr Opin Nephrol Hypertens 10: 321–329.
Springate JE, Feld LG, and Ganten D (1997). Enalapril and renal function in hypertensive rats transgenic for mouse renin gene. Hypertension 30: 868–872.
Terada Y, Tomita K, Homma MK, Nonoguchi H, Yang T, Yamada T, Yuasa Y, Krebs EG, Sasaki S, and Marumo F (1994). Sequential activation of Raf-1 kinase, mitogen-activated protein (MAP) kinase kinase, MAP kinase, and S6 kinase by hyperosmolality in renal cells. J Biol Chem 269: 31296–31301.
Tharaux PL, Chatziantoniou C, Fakhouri F, and Dussaule JC (2000). Angiotensin II activates collagen I gene through a mechanism involving the MAP/ER kinase pathway. Hypertension 36: 330–336.
Treisman R (1996). Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol 8: 205–215.
Underwood DC, Osborn RR, Bochnowicz S, Webb EF, Rieman DJ, Lee JC, Romanic AM, Adams JL, Hay DW, and Griswold DE (2000). SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lung. Am J Physiol Lung Cell Mol Physiol 279: L895–L902.
Ushio-Fukai M, Alexander RW, Akers M, and Griendling KK (1998). p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J Biol Chem 273: 15022–15029.
van Veldhuisen DJ, Boomsma F, de Kam PJ, Man in’t Veld AJ, Crijns HJ, Hampton JR, and Lie KI (1998). Influence of age on neurohormonal activation and prognosis in patients with chronic heart failure. Eur Heart J 19: 753–760.
Viedt C, Soto U, Krieger-Brauer HI, Fei J, Elsing C, Kubler W, and Kreuzer J (2000). Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II: Involvement of p22phox and reactive oxygen species. Arterioscler Thromb Vasc Biol 20: 940–948.
Witte K, Schnecko A, Schmidt T, Voll C, Kranzlin B, and Lemmer B (1999). Cardiovascular risk, renal hypertensive damage, and effects of amlodipine treatment in transgenic TGR(mREN2)27 rats. Gen Pharmacol 33: 423–430.
Wolf G, Haberstroh U, and Neilson EG (1992). Angiotensin II stimulates the proliferation and biosynthesis of type I collagen in cultured murine mesangial cells. Am J Pathol 140: 95–107.
Wolf G, Ziyadeh FN, Zahner G, and Stahl RA (1996). Angiotensin II is mitogenic for cultured rat glomerular endothelial cells. Hypertension 27: 897–905.
Wu LL, Cox A, Roe CJ, Dziadek M, Cooper ME, and Gilbert RE (1997). Transforming growth factor beta 1 and renal injury following subtotal nephrectomy in the rat: Role of the renin-angiotensin system. Kidney Int 51: 1553–1567.
Acknowledgements
The authors gratefully thank Dr. U. Ganten (Max Delbrück Center for Molecular Medicine, Berlin-Buch, Germany) for the generous gift of homozygous TGR(mRen2)27 rats, and Dr. R. N. Willette (Department of Cardiovascular Pharmacology, GlaxoSmithKline, King of Prussia, Pennsylvania) for the kind gift of the p38 MAP-kinase inhibitor SB239063.
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported by a grant from the Groningen University Institute for Drug Exploration (GUIDE).
Rights and permissions
About this article
Cite this article
de Borst, M., Navis, G., de Boer, R. et al. Specific MAP-Kinase Blockade Protects against Renal Damage in Homozygous TGR(mRen2)27 Rats. Lab Invest 83, 1761–1770 (2003). https://doi.org/10.1097/01.LAB.0000101731.11015.F6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.LAB.0000101731.11015.F6
This article is cited by
-
Possible renoprotection by vitamin D in chronic renal disease: beyond mineral metabolism
Nature Reviews Nephrology (2009)
-
Pharmacological Modulation of Epithelial Mesenchymal Transition Caused by Angiotensin II. Role of ROCK and MAPK Pathways
Pharmaceutical Research (2008)
-
Renal p38 MAP kinase activity in experimental diabetes
Laboratory Investigation (2007)
-
Mannose-6-Phosphate/Insulin-Like Growth Factor-II Receptors may Represent a Target for the Selective Delivery of Mycophenolic Acid to Fibrogenic Cells
Pharmaceutical Research (2006)
-
Calcineurin independent development of myocardial hypertrophy in transgenic rats overexpressing the mouse renin gene, TGR(mREN2)27
Journal of Molecular Medicine (2004)
