
Abstract
Because the mechanisms of telomerase activation in prostate cancer are mainly unknown, we investigated the relationships between telomerase activity and expression levels of human telomerase RNA (hTR) and human telomerase reverse transcriptase (hTERT) mRNA in benign and malignant alterations of the human prostate gland. Using the LightCycler technology, hTERT mRNA expression was quantified in 46 radical prostatectomy and 10 benign prostatic hyperplasia (BPH) cases; hTR expression was quantified in a subset of these tissue samples. Telomerase activity was measured using a quantitative telomeric repeat amplification protocol ELISA assay. Similar to hTR, which was expressed in all tissue samples tested, hTERT mRNA was detected in 98% of the prostate cancer samples and in 30% of the BPH samples. Regarding clinicopathologic variables, telomerase activity was significantly correlated with Gleason score (<7 vs ≥7, p = 0.02). No relationships emerged between normalized hTR or hTERT expression levels and tumor stage, Gleason score, lymph node status, or preoperative serum prostate-specific antigen. Remarkably, one third of all cancer and BPH tissue samples with hTR and hTERT expression lack telomerase activity. Quantitative analyses contradict the assumption that a certain threshold level of hTR or hTERT mRNA is required for telomerase activation, thus indicating that telomerase regulation in prostate cancer occurs more likely on a posttranscriptional level. Nevertheless, the observation that hTR and hTERT mRNA levels are significantly (p < 0.002) correlated suggests some common mechanisms in the up-regulation of hTR and hTERT expression. Because in situ hybridization revealed strong hTERT expression in all cells of the tumor glands but also in high-grade prostatic intraepithelial neoplasia foci, this up-regulation seems to occur early in prostate carcinogenesis.
Similar content being viewed by others


Introduction
Eukaryotic chromosomes are made up of an array of tandem repeats of the hexameric DNA sequence 5′-TTAGGG-3′, called telomeres. They protect the end of the chromosomes against degradation, end-to-end fusion, rearrangements, and loss of terminal coding DNA sequences (Zakian, 1989). With each cell division, these noncoding strands shorten by approximately 100 base pairs because of the inability of the conventional DNA polymerase to replicate 3′-terminal sequences (“end replication problem”) (Levy et al, 1992). The telomere hypothesis of cellular senescence postulates that progressive shortening of the chromosome ends results in cell cycle exit (Greider, 1990). Telomere length has been considered to function as a cellular replicative clock that limits replicative lifespan of normal somatic cells (Allsopp et al, 1992). In contrast to somatic cells, germ-line cells and proliferative cells of renewal tissue do not show telomere-shortening with increased age (Allsopp et al, 1992). This phenomenon is a result of the presence of the highly conserved ribonucleoprotein telomerase in these cells, synthesizing telomere repeats. Reactivation of telomerase has also been found in many human malignancies, suggesting that telomerase activation plays a crucial role in sustaining malignant tumor growth (Counter et al, 1994). Normal somatic human tissue predominantly lacks telomerase activity.
Human telomerase consists of two major components: human telomerase RNA (hTR), which provides the template for the synthesis of the human telomeric repeat (Feng et al, 1995), and human telomerase reverse transcriptase (hTERT), which provides catalytic function to replicate the ends of linear DNA (Weinrich et al, 1997). The presence of both subunits is mandatory for telomerase activity. Although hTR is constitutively expressed in all tissues, hTERT expression seems to be restricted to telomerase-positive tissue, indicating that hTERT is the limiting factor for telomerase activity (Bodnar et al, 1998; Counter et al, 1998; Weinrich et al, 1997). It should be mentioned that telomere stabilization can also be achieved by mechanisms that are independent of telomerase. These mechanisms, referred to as alternate lengthening of telomeres, have not been elucidated yet (Bryan et al, 1997).
(Re-)Activation of telomerase activity in most human cancers outlines the potential use of measuring telomerase activity or its subunits as a general molecular marker for malignant tumors. Moreover, it has been demonstrated for various tumors, including urothelial cell carcinoma (De Kok et al, 2000), colorectal carcinoma (Gertler et al, 2002), lung carcinoma (Wang et al, 2002) and breast cancer (Poremba et al, 2002), that assessment of hTERT expression levels may be used as prognostic marker.
In prostate cancer, the clinical relevance of the telomerase system is far less clear. Although telomerase activation is a frequent finding in prostate cancer, having been described in 58% to 93% of the tumors analyzed (Koeneman et al, 1998; Lin et al, 1997, 1998; Scates et al, 1997; Sommerfeld et al, 1996; Takahashi et al, 1997; Wullich et al, 1999; Wymenga et al, 2000; Zhang et al, 1998), it does not allow satisfactory distinction between benign and malignant hyperproliferation in the prostate gland. The reports on telomerase activation in normal prostate tissue and benign prostatic hyperplasia (BPH) are divergent, varying between 0% (Koeneman et al, 1998; Lin et al, 1997, 1998; Scates et al, 1997; Sommerfeld et al, 1996) and 50% (Wullich et al, 1999; Zhang et al, 1998). This warrants better knowledge of the telomerase regulatory mechanisms, which are mainly unknown in prostate cancer. One possible mechanism is transcriptional regulation of telomerase via expression of hTR and hTERT mRNA. To the best of our knowledge, there are only two studies analyzing hTERT expression in prostate cancer (Latil et al, 2000; Liu et al, 2001). Unfortunately, neither study gives any data on telomerase activity status, leaving the question about the relationship of hTERT expression and telomerase activity open. We therefore set out to quantify the expression of hTERT mRNA in 46 prostate cancer and 10 BPH cases and the expression of hTR in a subset of these tissue samples by applying real-time quantitative RT-PCR assays based on the LightCycler system. Telomerase activity was measured with a quantitative TRAP (telomeric repeat amplification protocol) ELISA assay. To determine the origin of hTERT mRNA expression at the cellular level, in situ hybridization was performed on normal prostate tissue, BPH, and prostate cancer.
Results
Telomerase Activity
Using the semiquantitative telomerase PCR ELISA assay, telomerase activity was detected in 32 (69.6%) of the 46 prostate carcinomas tested. In the remaining, telomerase activity was below the threshold value of [A450 nm − A690 nm] = 0.2. Of the 10 BPH probes, only one (10%) revealed weak telomerase activity. Regarding clinicopathologic parameters, telomerase activity was significantly correlated with the Gleason score (<7 vs ≥7; p = 0.02, Mann-Whitney U test; p = 0.008, Fisher's exact test) but not with tumor stage, lymph node status, nor preoperative serum prostate-specific antigen (PSA) (Fig. 1).

Distribution of telomerase activity levels in prostate cancer according to Gleason score. Samples have been regarded as telomerase negative if the difference in absorbance (A450 nm − A690 nm) of sample and negative control was <0.2 (marked by horizontal line). Significant correlation was documented only to Gleason score (<7 vs ≥7; p = 0.02, Mann-Whitney U test; p = 0.008, Fisher's exact test), not to tumor stage or lymph node status.
hTR and hTERT mRNA Expression
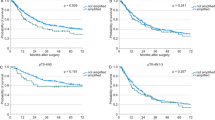
Using real-time quantitative RT-PCR based on the LightCycler technology, hTERT mRNA was detectable in 45 (97.8%) of the 46 prostate tumors tested. Measurement for hTR expression was possible on only 26 prostate tumors, because not enough material was available from all cases. Of these, hTR expression was detected in all samples. The median values of normalized hTERT mRNA and hTR were 0.04 (0–21.11) and 34.19 (0.05–3203.35), respectively. All samples showed correctly amplified porphobilinogen desaminase (PBGD) transcript, which was evaluated for internal PCR control. Of the telomerase-positive samples, all revealed detectable hTERT expression. Conversely, hTERT expression in tumors without telomerase activity was detected in 31.1%. Statistical analyses could only detect a relationship between normalized hTR and hTERT mRNA levels (p < 0.002) but not between normalized hTR or hTERT mRNA levels and telomerase measurements (Fig. 2). Regarding clinicopathologic variables, no statistically significant relationship between hTR or hTERT expression levels and tumor stage, Gleason score, lymph node status, or preoperative serum PSA emerged (Fig. 3).

Relationship among continuous variables such as telomerase activity, human telomerase reverse transcriptase (hTERT) mRNA, and human telomerase RNA (hTR) expression in prostate cancer. Associations were measured by Spearman's rank-order coefficient (rho). No correlation could be documented between telomerase activity and log(hTERT+0.1) (A) and between telomerase activity and log(hTR+0.1) (B); however, log(hTR+0.1) and log(hTERT+0.1) (C) were significantly correlated (p < 0.002). hTR and hTERT mRNA expression was normalized to porphobilinogen desaminase for each probe.

Distribution of hTERT expression level in prostate cancer according to Gleason score (A) and tumor stage (B). Neither Gleason score nor tumor stage correlated to hTERT expression level.
In BPH, three (30%) of the 10 samples had detectable hTERT expression and hTR was expressed in all (100%). Only one of the three hTERT-positive cases revealed telomerase activity. The median value of hTERT mRNA was 0.0063 (range, 0–0.0098).
hTERT in Situ Hybridization
In prostate cancer, hTERT signals were detected in all tumor glands, with the signals showing a homogeneous distribution pattern (Fig. 4). There was no obvious difference of staining intensity comparing tumor areas of different Gleason grade. At the cellular level, positive staining of hTERT was localized almost exclusively in the cytoplasm. Interestingly, in the high-grade prostatic intraepithelial neoplasia (PIN) foci, which were observed in the area of the cancer, hTERT expression was also detected in the epithelial compartment at a similar level to that in carcinoma cells. Although surrounding stromal cells did not show staining for hTERT, endothelial cells and inflammatory cells were almost all hTERT positive. Strong staining for hTERT also showed the mucosal cells of the prostatic urethra.

hTERT mRNA expression in benign, dysplastic, and malignant prostate tissue using in situ hybridization. A, Normal prostate glands. Labeling is restricted to the basal cell layer (magnification, × 200). No labeling is observed in the secretory cells and stromal tissue. B, High-grade prostatic intraepithelial neoplasia (PIN) lesion. All epithelial cells of PIN lesions express hTERT mRNA at a high level (magnification, × 200). C, Primary prostate cancer, Gleason 3. Strong labeling is observed in all tumor glands revealing a homogeneous distribution pattern. Note hTERT mRNA expression also in the endothelial cells of the tumor microvessels (arrows). Again, stromal tissue is unlabeled (magnification, × 100; inset × 200).
In normal prostate tissue and BPH, hTERT expression was observed in normal glands. In these glands, expression was restricted to the basal cell layer (Fig. 4). No expression was detected either in the epithelial secretory cells or in stromal tissue. In glands with basal cell hyperplasia, the cells revealed a homogeneous staining pattern. Specificity of the staining signals was confirmed by hybridization, with the sense probe showing no staining.
Discussion
Because little is known about how telomerase activity is regulated in prostate cancer, we investigated the relationship between telomerase activity and the level of hTR or hTERT mRNA expression in normal and malignant prostate tissue samples. Concerning telomerase activity in prostate cancer, the results of this study are in accordance with data published by others (Koeneman et al, 1998; Sommerfeld et al, 1996; Takahashi et al, 1997; Zhang et al, 1998). We detected telomerase activity in 70% of the prostate carcinoma samples tested. In contrast to our previous findings on radical prostatectomy specimens (Wullich et al, 1999), the current series revealed a significant correlation between telomerase activity and Gleason score. No correlation was detected to tumor stage, lymph node status, and preoperative PSA. Concerning the correlation between telomerase activity and Gleason score, the divergent results of our two studies might be explained by our previously reported observation of focal intratumoral heterogeneity for telomerase activity in prostate cancer. Because heterogeneity was more pronounced in better differentiated tumors, testing of only a single tissue sample per case, as was done in the current study, may lead to a higher detection rate of telomerase-negativity, particularly in tumors with a lower Gleason score.
In the 10 BPH samples, telomerase activity was found in only one sample (10%). Including our recently published series on eight BPH samples (Wullich et al, 1999), the overall frequency of telomerase activity in BPH was 16.7%.
Focusing on the major components of the telomerase complex, we performed quantitative measurements of hTR and hTERT mRNA expression in prostate cancer. By quantitative real-time RT-PCR using the LightCycler technology, hTERT expression was detected in 45 (98%) of the 46 prostate cancers samples analyzed. Remarkably, 14 of the 45 cancer samples with hTERT expression lacked telomerase activity, contradicting the assumption that hTERT positivity is a predictor of telomerase activity. One explanation for this discrepancy could be the existence of a certain threshold level of hTERT mRNA that is required for telomerase activation (Hisatomi et al, 1999; Snijders et al, 1998). We indeed observed a bright range of hTERT expression levels (normalized to PDBG) in the prostate carcinomas analyzed; however, no correlation could be documented between telomerase positivity and level of hTERT expression. Unfortunately, no autologous prostate tissue without tumor infiltration was available for this investigation, so that we do not know whether there are specific baseline expression levels in individual carcinomatous prostates. Nevertheless, our findings on BPH samples from control patients without prostate cancer demonstrate that low hTERT expression can also be detected in noncancerous tissue samples lacking telomerase activity.
Quantitative expression of hTERT in prostate cancer was also studied by Latil et al (2000). Of the 33 tumors analyzed, 67% had a detectable amount of hTERT mRNA. In accordance with our findings, they reported a high variation of hTERT expression levels. Unfortunately, the telomerase activity status of the tumors was not determined. The difference in the frequency of hTERT positivity between the current series and the one of Latil et al (2000) could be explained by different real-time RT-PCR techniques or sampling bias. Good validation of the kit we used is documented by others (Schneider-Stock et al, 2001; Yi et al, 2001). With regard to sampling, the study of Latil et al (2000) also included tissue samples from patients who received androgen ablation therapy before radical prostatectomy. The effect of androgen depletion on hTERT expression is still unknown.
In contrast to our own observations, various studies have described a close relationship between hTERT expression and telomerase activity. It has been recommended to use quantitative measurement of hTERT as an alternative to time-consuming telomerase activity measurement by TRAP assay (Kyo et al, 1999; Park et al, 1999; Wu et al, 1999). This suggestion, however, has to be questioned at least for prostate tissue; there is increasing evidence that similar discrepancies between level of hTERT expression and telomerase activity also exist in other tissue types, either normal or neoplastic (Nakamura et al, 1999; Rohde et al, 2000; Tahara et al, 1999; Ulaner et al, 2000). In vitro studies consistently indicate other factors in addition to hTERT mRNA that recruit and regulate telomerase (McChesney et al, 2000; Yi et al, 2001).
With regard to clinical implications of hTERT mRNA expression, our data do not show any relationship between hTERT expression level and tumor stage, Gleason score, lymph node status, and preoperative PSA. These results agree with the study of Latil et al (2000), which indicate that hTERT expression is not an index of malignant potential of prostate cancer. However, the mean follow-up in our study was only 2 years, whereas prospective studies lasting several years will be necessary to determine the prognostic value of hTERT in prostate cancer. Considering other solid tumors, investigations on gastrointestinal (Nakamura et al, 1999) and cervical tumors (Wisman et al, 2001) yielded similar results demonstrating that hTERT was not applicable as a prognostic factor. This, however, contrasts with reports demonstrating high expression levels of hTERT to be correlated with aggressiveness of breast cancer (Bieche et al, 2000; Poremba et al, 2002), liposarcoma (Schneider-Stock et al, 2001), non-small cell lung cancer (Wang et al, 2002), and colorectal cancer (Gertler et al, 2002) and with recurrence risk of Wilms tumors (Dome et al, 1999).
In accordance with observations that the hTR gene is constitutively expressed (Avilion et al, 1996; Feng et al, 1995), our analyses revealed hTR expression in all tumor and BPH samples tested. Expression of hTR in 100% of the normal and malignant prostate tissue samples analyzed was also reported by another study using conventional RT-PCR (Liu et al, 2001). A lower detection rate was reported in an in situ hybridization study, yielding hTR expression in 75% of the nine prostate tumors analyzed (Paradis et al, 1999). They observed hTR expression in the carcinomatous glands but also in the epithelial cells of high-grade PIN foci and, notably, in the basal cells of normal prostate tissue. Based on these findings, we wondered whether hTERT mRNA expression displays a similar distribution pattern at the cellular level in normal, dysplastic, and cancerous prostate tissue. Using in situ hybridization, we demonstrated strong hTERT expression in the tumor glands. No difference in the signal intensities could be observed between low- and high-grade tumor lesions. In analogy to hTR expression, we also recognized strong expression of hTERT mRNA in the epithelial cells of high-grade PIN foci. Because it is generally accepted that high-grade PIN is a precursor of prostate cancer, this finding indicates that hTERT is expressed early in prostate carcinogenesis.
One interesting finding in our study concerns hTERT expression in normal prostate tissue and BPH. This labeling was restricted to the basal cells of normal glands. The secretory luminal cells were hTERT negative, supporting experimental models that have shown basal cells to behave as stem cells that differentiate to secretory cells (Bonkhoff and Remberger, 1996). The low number of hTERT-expressing cells in the whole tissue of the BPH samples corresponds to the low overall levels of hTERT mRNA, which were measured in BPH by real-time RT-PCR.
Although not unexpected, the concordance of hTR and hTERT expression pattern in normal prostate glands and prostate cancer tissue is remarkable, indicating some common mechanisms in the up-regulation of hTR and hTERT expression in prostate cells. This observation is confirmed by immunohistochemical detection of the hTERT protein in human cancer and some normal tissues, which reveals many cells with only hTR signals, but no cells with hTERT signals without hTR expression (Hiyama et al, 2001). Cell culture experiments further indicate an overlap in the transcriptional regulatory control of the two genes (Yi et al, 1999).
The observation that hTR- and hTERT-expressing normal and neoplastic prostate tissue samples may lack telomerase activity allows two possible interpretations. First, the level of telomerase activity is too low to be detected by the TRAP assay. This is reasonable in normal prostate tissue or BPH in which only a minority of cells, namely the basal cells, contribute to the overall level of telomerase activity. In tumor samples, however, telomerase activation would hardly be missed by the TRAP assay. An intermediate status can be imagined for certain benign alterations that are associated with either a numerical expansion of hTERT-positive cells, such as in basal cell hyperplasia, or with other hyperproliferative conditions, such as BPH. This might explain why a non-negligible proportion of BPH has been described to reveal telomerase activity (Wullich et al, 1999; Wymenga et al, 2000). Second, expression of hTR and hTERT alone are not sufficient to determine telomerase activity. Other mechanisms, such as posttranscriptional modification of hTERT (Akalin et al, 2001; Kilian et al, 1997; Ulaner et al, 2000; Yi et al, 2000) or inactivation of inhibitors of telomerase (Horikawa et al, 1998; Nishimoto et al, 2001), may be involved in the acquisition of enzymatic activity. Organ-specific differences in the regulation of telomerase activity are likely.
Compared with hTERT, measurement of telomerase activity, although no tumor marker, seems to better predict the biologic properties of prostate cancer. It would be interesting to analyze whether determination of telomere length, which is directly influenced by the level of telomerase activity, may refine the value of telomerase activity as a prognostic tool. In addition to telomeric restriction fragment length analysis, novel methods using fluorescence in situ hybridization techniques (telomere fluorescence in situ hybridization) can now be applied (Scherthan, 2002).
Materials and Methods
Tissue Samples
All cancer tissue samples were collected from patients (n = 46) who underwent radical prostatectomy for prostate carcinoma between 1999 and 2001. Histologic diagnosis was performed on the same samples that were used for this study. To minimize sampling error, control sections from the top and the bottom of each sample were examined using standard staining protocols. Only samples containing at least 70% tumor cells in both sections were used for further studies. Tumor staging ranged from pT1b to pT4: 1 × pT1b, 1 × pT2a, 8 × pT2b, 16 × pT3a, 14 × pT3b, 2 × pT3c, and 4 × pT4. Distribution of the Gleason scores was as follows: 4 × <7, 27 × = 7, 15 × > 7. In seven cases, lymph node metastases (pN1) were diagnosed. None of the patients had been treated with radiation or androgen depletion before operation. For control, BPH probes were obtained from patients (n = 10) who were treated by transurethral resection because of obstructive voiding disease.
RNA and Protein Extraction
Total RNA was isolated from frozen tissue using TriZol reagent (Life Technologies, Eggenstein, Germany). RNA purification and Dnase digestion were performed using the StrataPrep Total RNA Miniprep Kit (Stratagene, La Jolla, California) according to the manufacturer's applications. Isolated RNA was stored at −70° C for further use.
Protein was extracted using lysis reagent of the TeloTAGGG Telomerase PCR ELISA (Roche Diagnostics, Mannheim, Germany). Briefly, 70 mg of frozen tissue was homogenized in ice-cold lysis buffer, incubated on ice for 30 minutes, and then centrifuged. Supernatant was carefully removed and stored at −70° C for further use.
TRAP Assay
For detection of telomerase activity, a photometric enzyme immunoassay using the telomeric repeat amplification protocol was used (TeloTAGGG Telomerase PCR ELISA; Roche). The test was performed according to the manufacturer's instructions. Briefly, 1 μl of tissue extract (8 μg total protein) was used for combined primer elongation/amplification reaction. Five microliters of the amplification product was transferred for hybridization and the ELISA procedure. ELISA was analyzed within 30 minutes after the addition of stop reagent with a photometer (Ultrospec 3000; Pharmacia Biotech, Cambridge, United Kingdom) at 450 nm and a 690 nm reference wavelength. Each assay contained a negative and positive control (protein extract of PC3 cell line). Results of the assay were accepted only if negative controls had an absorbance (A450 nm − A690 nm) less 0.25. Samples were regarded as telomerase positive if the difference in absorbance of sample and negative control was higher than 0.2. All measurements were performed twice to ensure reliability of the test. To exclude the presence of Taq polymerase inhibitors in the tissue extracts of the telomerase-negative samples, they were checked by another TRAP assay (TeloTAGGG PCR ELISA plus; Roche), including a 216-bp internal DNA standard.
Real-Time RT-PCR
Measurements of hTR and hTERT mRNA expression were performed on a LightCycler instrument (Roche). One-step RT-PCR kits TeloTAGGG hTR and TeloTAGGG hTERT (Roche) were used for the quantification of hTR and hTERT mRNA content, respectively. Both kits use hybridization probe pairs for a highly specific detection of the amplicons. These probes hybridize to an internal segment of the amplified fragment. One is labeled with LightCycler Red-640 at the 5′ end and the other with fluorescein at the 3′ end. If both probes hybridize to the template DNA in close proximity, then fluorescence resonance energy transfer will occur with fluorescence emission of LightCycler Red-640, which is measured with the LightCycler instrument. In a separate RT-PCR, mRNA encoding PBGD was processed. PBGD expression served for RT-PCR performance control and for relative quantification.
Real-time RT-PCR was performed according to the manufacturer's instructions. Briefly, 150 ng of total RNA was used per reaction. Positive and negative controls and five internal standard solutions were added to each LightCycler run. Because the hTR-encoding gene is an intron-free gene, controls without the reverse transcriptase were added to each RT-PCR run to ensure no DNA contamination. According to the manufacturer's specifications, primers for hTERT and PBGD span exon-intron boundaries, preventing coamplification of DNA.
Quantitative analysis was achieved using LighCycler software 3.5. Data of a PCR run were only accepted if the regression coefficient of internal standards was r = −1 and calculated error was ≤0.19. The copy number of hTR, hTERT mRNA, and PBGD mRNA in each reaction was computed by LightCycler software using the second derivative maximum method. Normalization of hTR and hTERT mRNA expression data for sample to sample differences in RNA input, RNA quality, and RT efficiency was achieved by dividing copy numbers of hTERT mRNA and hTR with copy numbers of PBGD mRNA for each probe. Identity and specificity of PCR products were confirmed by melting curve analysis, which is part of the LightCycler software.
hTERT in Situ Hybridization
For hTERT detection, a previously described 1261-bp cDNA fragment (from nucleotides 1895–3155) cloned into a PCR II vector was used as probe. The 1261-bp fragment was a kind gift from Dr. Nan-Ping Weng (Laboratory of Immunology, Gerontology Research Center, National Institute on Aging, National Institutes of Health, Baltimore, Maryland) (Liu et al, 1999). Sense and antisense hTERT probes were synthesized by in vitro transcription using the T7 Transcription Kit (MBI Fermentas, St. Leon-Roth, Germany) and labeled with digoxigenin-dUTP (Roche). The sense probe served for negative control. Formalin-fixed, paraffin-embedded tissue sections (5-μm thickness) were deparaffinized, and after digestion in 400 μg/ml proteinase K (Merck, Darmstadt, Germany) at 37° C for 15 minutes, the sections were fixed with 4% paraformaldehyde in PBS for 5 minutes at room temperature. After they were washed twice in 2 × SSC (Merck), the slides were covered with 40 μl of the prehybridization buffer containing 2 × SSC, 1 × Denhardt's solution (50 × Denhardt's stock solution: 1% polyvinylchloride, 1% pyrrolidone, 2% BSA; Oncor, Heidelberg, Germany), 10% dextran sulfate (Roche Diagnostics), 50 mm phosphate buffer (pH 7.0; Merck), 50 mm 1,4-dithiothreitol (Roche Diagnostics), 250 μg/ml yeast tRNA (Roche Diagnostics), 100 μg/ml polyadenylic acid (Roche Diagnostics), 500 μg/ml denatured and sheared DNA from fish sperm (Roche Diagnostics), and 40% deionized formamide (Oncor); slides were then incubated for 2 hours at 41° C in a humid chamber. The sections were hybridized overnight at 41° C with 40 μl of hybridization mixture containing the hybridization buffer and 10 pm labeled hTERT probe per slide. After washing in graded concentrations of SSC at 37° C for 30 minutes, the sections were preincubated with 2% BSA solution for 20 minutes and then incubated with antiDig-POD FAB fragments (Roche). A signal amplification method based on the deposition of biotinylated tyramine was used to enhance immunodetection. After precipitation of the biotinylated tyramine (10 minutes at room temperature) through the enzymatic action of horseradish peroxidase (HRP) and H2O2 (0.1%), the biotin precipitate was detected with an additional application of the HRP-labeled avidin-biotin complex (ABComplex/HRP; Dako, Hamburg, Germany) for 30 minutes in a humid chamber. The peroxidase reaction was developed by 3,3′-diaminobenzidine (Sigma, Deisenhofen, Germany).
In each experiment, an RNase A-digested slide was used as negative control. The slides were digested for 30 minutes at 37° C in a digestion buffer (0.5 m NaCl, 10 mm Tris-HCl, 1 mm EDTA, pH 7.2) containing 10 μg/μl RNase A (Roche Diagnostics). Only RNase-sensitive signals were considered positive for hTERT mRNA expression.
Statistical Analysis
As a measure of association for nominal data (telomerase activity, yes/no; Gleason score, <7 vs ≥7; organ-confined (pT1/2) vs extracapsular (pT3/4) growth; and lymph node status, yes/no), Fisher's exact test was applied (Feldman and Klinger, 1963). Because of the right skewness of the distribution of hTERT and hTR normalized levels, these continuous variables were transformed by taking their logarithms. The value of 0.1 was added to all records as performed elsewhere (Dome et al, 1999) to avoid an infinite logarithm of those samples with a hTERT or hTR level of zero. Relationships between continuous variables were assessed by Spearman's rank-order coefficient (Spearman, 1904), because most variables did not comply with normal Gaussian distribution (Shapiro and Wilk, 1965). To compare independent samples of continuous variables grouped by nominal or ordinal data, the nonparametric Mann-Whitney U test or Kruskal-Wallis test was performed, respectively (Kruskal and Wallis, 1952; Mann and Whitney, 1947). All p values were based on two-sided tests, and the threshold to accept statistical significance was set at the alpha level 0.05. Analyses were performed with the statistical software package SPSS version 10.0 (SPSS Inc., Chicago, Illinois).
References
Akalin A, Elmore LW, Forsythe HL, Amaker BA, McCollum ED, Nelson PS, Ware JL, and Holt SE (2001). A novel mechanism for chaperone-mediated telomerase regulation during prostate cancer progression. Cancer Res 61: 4791–4796.
Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, Greider CW, and Harley CB (1992). Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA 89: 10114–10118.
Avilion AA, Piatyszek MA, Gupta J, Shay JW, Bacchetti S, and Greider CW (1996). Human telomerase RNA and telomerase activity in immortal cell lines and tumor tissues. Cancer Res 56: 645–650.
Bieche I, Nogues C, Paradis V, Olivi M, Bedossa P, Lidereau R, and Vidaud M (2000). Quantitation of hTERT gene expression in sporadic breast tumors with a real-time reverse transcription-polymerase chain reaction assay. Clin Cancer Res 6: 452–459.
Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, and Wright WE (1998). Extension of life-span by introduction of telomerase into normal human cells. Science 279: 349–352.
Bonkhoff H and Remberger K (1996). Differentiation pathways and histogenetic aspects of normal and abnormal prostatic growth: A stem cell model. Prostate 28: 98–106.
Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, and Reddel RR (1997). Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med 3: 1271–1274.
Counter CM, Botelho FM, Wang P, Harley CB, and Bacchetti S (1994). Stabilization of short telomeres and telomerase activity accompany immortalization of Epstein-Barr virus-transformed human B lymphocytes. J Virol 68: 3410–3414.
Counter CM, Meyerson M, Eaton EN, Ellisen LW, Caddle SD, Haber DA, and Weinberg RA (1998). Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene 16: 1217–1222.
De Kok JB, Schalken JA, Aalders TW, Ruerst JM, Willems HL, and Swinkels DW (2000). Quantitative measurement of telomerase reverse transcriptase (hTERT) mRNA in urothelial cell carcinomas. Int J Cancer 87: 217–220.
Dome JS, Chung S, Bergemann T, Umbricht B, Saji M, Carey LA, Grundy PE, Perlman EJ, Breslow NE, and Sukumar S (1999). High telomerase reverse transcriptase (hTERT) messenger RNA level correlates with tumor recurrence in patients with favorable histology Wilms' tumor. Cancer Res 59: 4301–4307.
Feldman SE and Klinger E (1963). Short calculation of the Fisher-Yates “exact test.” Psychometrika 28: 289–291.
Feng J, Funk WD, Wang SS, Weinrich SL, Avilion AA, Chiu CP, Adams RR, Chang E, Allsopp RC, and Yu J (1995). The RNA component of human telomerase. Science 269: 1236–1241.
Gertler R, Rosenbaum R, Stricker D, Werner M, Lassmann S, Ulm K, Nekarda H, and Siewert JR (2002). Prognostic potential of the telomerase subunit human telomerase reverse transcriptase in tumor tissue and nontumorous mucosa from patients with colorectal carcinoma. Cancer 95: 2103–2111.
Greider CW (1990). Telomeres, telomerase and senescence. Bioessays 12: 363–369.
Hisatomi H, Nagao K, Kanamaru T, Endo H, Tomimatsu M, and Hikiji K (1999). Levels of telomerase catalytic subunit mRNA as a predictor of potential malignancy. Int J Oncol 14: 727–732.
Hiyama E, Hiyama K, Yokoyama T, and Shay JW (2001). Immunohistochemical detection of telomerase (hTERT) protein in human cancer tissues and a subset of cells in normal tissues. Neoplasia 3: 17–26.
Horikawa I, Oshimura M, and Barrett JC (1998). Repression of the telomerase catalytic subunit by a gene on human chromosome 3 that induces cellular senescence. Mol Carcinog 22: 65–72.
Kilian A, Bowtell DD, Abud HE, Hime GR, Venter DJ, Keese PK, Duncan EL, Reddel RR, and Jefferson RA (1997). Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet 6: 2011–2019.
Koeneman KS, Pan CX, Jin JK, Pyle JM, Flanigan RC, Shankey TV, and Diaz MO (1998). Telomerase activity, telomere length, and DNA ploidy in prostatic intraepithelial neoplasia (PIN). J Urol 160: 1533–1539.
Kruskal WH and Wallis WA (1952). Use of ranks in one-criterion variance analysis. J Am Stat Assoc 47: 583–621.
Kyo S, Kanaya T, Takakura M, Tanaka M, Yamashita A, Inoue H, and Inoue M (1999). Expression of human telomerase subunits in ovarian malignant, borderline and benign tumors. Int J Cancer 80: 804–809.
Latil A, Vidaud D, Valeri A, Fournier G, Vidaud M, Lidereau R, Cussenot O, and Biache I (2000). hTERT expression correlates with MYC over-expression in human prostate cancer. Int J Cancer 89: 172–176.
Levy MZ, Allsopp RC, Futcher AB, Greider CW, and Harley CB (1992). Telomere end-replication problem and cell aging. J Mol Biol 225: 951–960.
Lin Y, Uemura H, Fujinami K, Hosaka M, Harada M, and Kubota Y (1997). Telomerase activity in primary prostate cancer. J Urol 157: 1161–1165.
Lin Y, Uemura H, Fujinami K, Hosaka M, Iwasaki Y, Kitamura H, Harada M, and Kubota Y (1998). Detection of telomerase activity in prostate needle-biopsy samples. Prostate 36: 121–128.
Liu BC, LaRose I, Weinstein LJ, Ahn M, Weinstein MH, and Richie JP (2001). Expression of telomerase subunits in normal and neoplastic prostate epithelial cells isolated by laser capture microdissection. Cancer 92: 1943–1948.
Liu K, Schoonmaker MM, Levine BL, June CH, Hodes RJ, and Weng NP (1999). Constitutive and regulated expression of telomerase reverse transcriptase (hTERT) in human lymphocytes. Proc Natl Acad Sci USA 96: 5147–5152.
Mann HB and Withney DR (1947). On a test of whether one of two random variables is stochastically larger than the other. Ann Math Statist 18: 50–60.
McChesney PA, Aisner DL, Frank BC, Wright WE, and Shay JW (2000). Telomere dynamics in cells with introduced telomerase: A rapid assay for telomerase activity on telomeres. Mol Cell Biol Res Commun 3: 312–318.
Nakamura Y, Tahara E, Tahara H, Yasui W, and Ide T (1999). Quantitative reevaluation of telomerase activity in cancerous and noncancerous gastrointestinal tissues. Mol Carcinog 26: 312–320.
Nishimoto A, Miura N, Horikawa I, Kugoh H, Murakami Y, Hirohashi S, Kawasaki H, Gazdar AF, Shay JW, Barrett JC, and Oshimura M (2001). Functional evidence for a telomerase repressor gene on human chromosome 10p15.1. Oncogene 20: 828–835.
Paradis V, Dargere D, Laurendeau I, Benoit G, Vidaud M, Jardin A, and Bedossa P (1999). Expression of the RNA component of human telomerase (hTR) in prostate cancer, prostatic intraepithelial neoplasia, and normal prostate tissue. J Pathol 189: 213–218.
Park TW, Riethdorf S, Riethdorf L, Loning T, and Janicke F (1999). Differential telomerase activity, expression of the telomerase catalytic sub-unit and telomerase-RNA in ovarian tumors. Int J Cancer 84: 426–431.
Poremba C, Heine B, Diallo R, Heinecke A, Wai D, Schaefer KL, Braun Y, Schuck A, Lanvers C, Bankfalvi A, Kneif S, Torhorst J, Zuber M, Kochli OR, Mross F, Dieterich H, Sauter G, Stein H, Fogt F, and Boecker W (2002). Telomerase as a prognostic marker in breast cancer: High-throughput tissue microarray analysis of hTERT and hTR. J Pathol 198: 181–189.
Rohde V, Sattler HP, Bund T, Bonkhoff H, Fixemer T, Bachmann C, Lensch R, Unteregger G, Stoeckle M, and Wullich B (2000). Expression of the human telomerase reverse transcriptase is not related to telomerase activity in normal and malignant renal tissue. Clin Cancer Res 6: 4803–4809.
Scates DK, Muir GH, Venitt S, and Carmichael PL (1997). Detection of telomerase activity in human prostate: A diagnostic marker for prostatic cancer? Br J Urol 80: 263–268.
Scherthan H (2002). Detection of chromosome ends by telomere FISH. Methods Mol Biol 191: 13–31.
Schneider-Stock R, Emrich T, Peters B, Jaeger V, and Roessner A (2001). Analysis of human telomerase reverse transcriptase mRNA (hTERT) expression in myxoid liposarcomas using LightCycler real-time quantitative reverse transcriptase-polymerase chain reaction. Electrophoresis 22: 1098–1101.
Shapiro SS and Wilk MB (1965). An analysis of variance test for normality (complete samples). Biometrika 52: 591–611.
Snijders PJ, van Duin M, Walboomers JM, Steenbergen RD, Risse EK, Helmerhorst TJ, Verheijen RH, and Meijer CJ (1998). Telomerase activity exclusively in cervical carcinomas and a subset of cervical intraepithelial neoplasia grade III lesions: Strong association with elevated messenger RNA levels of its catalytic subunit and high-risk human papillomavirus DNA. Cancer Res 58: 3812–3818.
Sommerfeld HJ, Meeker AK, Piatyszek MA, Bova GS, Shay JW, and Coffey DS (1996). Telomerase activity: A prevalent marker of malignant human prostate tissue. Cancer Res 56: 218–222.
Spearman C (1904). The proof and measurement of association between two things. Am J Psychol 15: 72–101.
Tahara H, Yasui W, Tahara E, Fujimoto J, Ito K, Tamai K, Nakayama J, Ishikawa F, and Ide T (1999). Immuno-histochemical detection of human telomerase catalytic component, hTERT, in human colorectal tumor and non-tumor tissue sections. Oncogene 18: 1561–1567.
Takahashi C, Miyagawa I, Kumano S, and Oshimura M (1997). Detection of telomerase activity in prostate cancer by needle biopsy. Eur Urol 32: 494–498.
Ulaner GA, Hu JF, Vu TH, Oruganti H, Giudice LC, and Hoffman AR (2000). Regulation of telomerase by alternate splicing of human telomerase reverse transcriptase (hTERT) in normal and neoplastic ovary, endometrium and myometrium. Int J Cancer 85: 330–335.
Wang L, Soria JC, Kemp BL, Liu DD, Mao L, and Khuri FR (2002). hTERT expression is a prognostic factor of survival in patients with stage I non-small cell lung cancer. Clin Cancer Res 8: 2883–2889.
Weinrich SL, Pruzan R, Ma L, Ouellette M, Tesmer VM, Holt SE, Bodnar AG, Lichtsteiner S, Kim NW, Trager JB, Taylor RD, Carlos R, Andrews WH, Wright WE, Shay JW, Harley CB, and Morin GB (1997). Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat Genet 17: 498–502.
Wisman GB, Knol AJ, Helder MN, Krans M, de Vries EG, Hollema H, de Jong S, and van der Zee AG (2001). Telomerase in relation to clinicopathologic prognostic factors and survival in cervical cancer. Int J Cancer 91: 658–664.
Wu A, Ichihashi M, and Ueda M (1999). Correlation of the expression of human telomerase subunits with telomerase activity in normal skin and skin tumors. Cancer 86: 2038–2044.
Wullich B, Rohde V, Oehlenschlager B, Bonkhoff H, Ketter R, Zwergel T, and Sattler HP (1999). Focal intratumoral heterogeneity for telomerase activity in human prostate cancer. J Urol 161: 1997–2001.
Wymenga LF, Wisman GB, Veenstra R, Ruiters MH, and Mensink HJ (2000). Telomerase activity in needle biopsies from prostate cancer and benign prostates. Eur J Clin Invest 30: 330–335.
Yi X, Shay JW, and Wright WE (2001). Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic Acids Res 29: 4818–4825.
Yi X, Tesmer VM, Savre-Train I, Shay JW, and Wright WE (1999). Both transcriptional and posttranscriptional mechanisms regulate human telomerase template RNA levels. Mol Cell Biol 19: 3989–3997.
Yi X, White DM, Aisner DL, Baur JA, Wright WE, and Shay JW (2000). An alternate splicing variant of the human telomerase catalytic subunit inhibits telomerase activity. Neoplasia 2: 433–440.
Zakian VA (1989). Structure and function of telomeres. Annu Rev Genet 23: 579–604.
Zhang W, Kapusta LR, Slingerland JM, and Klotz LH (1998). Telomerase activity in prostate cancer, prostatic intraepithelial neoplasia, and benign prostatic epithelium. Cancer Res 58: 619–621.
Acknowledgements
The study was supported by the Wilhelm-Sander-Stiftung (Grant 98.105.1 to BW).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kamradt, J., Drosse, C., Kalkbrenner, S. et al. Telomerase Activity and Telomerase Subunit Gene Expression Levels Are Not Related in Prostate Cancer: A Real-Time Quantification and In Situ Hybridization Study. Lab Invest 83, 623–633 (2003). https://doi.org/10.1097/01.LAB.0000069035.85309.30
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.LAB.0000069035.85309.30
This article is cited by
-
Expression of two non-mutated genetic elements is sufficient to stimulate oncogenic transformation of human mammary epithelial cells
Cell Death & Disease (2018)
-
DKC1 overexpression associated with prostate cancer progression
British Journal of Cancer (2009)
