
Abstract
Since the first report of long-chain L-3-hydroxyacyl-coenzyme A dehydrogenase deficiency a little more than a decade ago, its phenotypic and genotypic heterogeneity in individuals homozygous for the enzyme defect has become more and more evident. Even more interesting is its association with pregnancy-specific disorders, including preeclampsia, HELLP syndrome (hemolysis, elevated liver enzymes, low platelets), hyperemesis gravidarum, acute fatty liver of pregnancy, and maternal floor infarct of the placenta. In this review we discuss the biochemical and molecular basis, clinical features, diagnosis, and management of long-chain L-3-hydroxyacyl-coenzyme A dehydrogenase deficiency.
Similar content being viewed by others


Introduction
Long-chain L-3-hydroxyacyl-coenzyme A dehydrogenase (LCHAD) is an enzyme involved in the β-oxidation of long-chain fatty acids and has been under scientific scrutiny recently because of its reported association with certain pregnancy-specific disorders. Schoeman et al (1991) first suggested a link between recurrent acute fatty liver of pregnancy (AFLP) and a defect in fatty acid oxidation. Since then, more evidence has accumulated to suggest that defects in fatty acid oxidation, especially deficiency of LCHAD, may be etiologically related to pregnancy-specific diseases, including preeclampsia, HELLP syndrome (hemolysis, elevated liver enzymes, low platelets), hyperemesis gravidarum, maternal floor infarct of the placenta, and AFLP. Wanders et al (1989) published the first case of biochemically proven LCHAD deficiency in a child, and the clinical profile of this metabolic disorder continues to evolve as more cases come to light.
Herein we review the β-oxidation of fatty acids and the molecular basis of LCHAD deficiency, followed by clinical features, diagnosis, and management of this disorder.
Long-Chain Fatty Acid Oxidation
Fatty acid catabolism may involve α-, β-, or ω-oxidation, but most fatty acids undergo β-oxidation that in higher eukaryotes can occur both in mitochondria and in peroxisomes (Wanders et al, 1999). Depending on the number of carbon atoms, there are four arbitrarily defined groups of fatty acids: short-chain fatty acids containing 2 to 4 carbon atoms, medium-chain fatty acids containing 6 to 10 carbon atoms, long-chain fatty acids containing 12 to 18 carbon atoms, and very long-chain fatty acids containing 20 to 26 carbon atoms. The long-chain fatty acids containing an even number of carbon atoms are important human nutrients (Rifai et al, 2001), with palmitic acid (16 carbon atoms: 0 double bonds or 16:0), stearic acid (18:0), oleic acid (18 carbon atoms: 1 double bond at position 9 from the methyl end or 18:1ω9), and linoleic acid (18 carbon atoms: 2 double bonds, with the double bond nearest to the methyl end at position 6 or 18:2ω6) together accounting for more than 90% of the fatty acids in the U.S. diet (Veldee, 2001).
For mitochondrial β-oxidation of long-chain fatty acids to occur, the fatty acids have to first enter the mitochondria across the selectively permeable mitochondrial membranes (Fig. 1). Free fatty acids are activated to their respective coenzyme A (CoA) esters at the outer mitochondrial membrane by ATP-dependent acyl-CoA synthetases (Bennett et al, 2000):

Transport of long-chain free fatty acids into mitochondria.
R-COOH + CoASH + ATP → R-CO-SCoA + AMP + PiPi + H2O
Acyl-CoA synthetases also transport the acyl-CoAs into the mitochondrial intermembrane space. From there, the acyl-CoAs are transported into the inner mitochondrial matrix via the carnitine shuttle (McGarry and Brown, 1997). The enzyme carnitine-palmitoyl transferase 1 (CPT1) present on the inner aspect of the outer mitochondrial membrane converts acyl-CoA into an acyl-carnitine, using up free carnitine (β-hydroxy γ-trimethylaminobutyric acid) in the process:
R-CO-SCoA + carnitine → R-CO-carnitine + CoA
The acyl-carnitine crosses the inner mitochondrial membrane in exchange for free carnitine via the transport protein, carnitine-acylcarnitine translocase, located in the inner mitochondrial membrane. The enzyme carnitine-palmitoyl transferase 2 (CPT2), present on the inner aspect of the inner mitochondrial membrane, regenerates acyl-CoA–free carnitine from acyl-carnitine and free coenzyme A:
R-CO-carnitine + CoA ←→ R-CO-SCoA + carnitine
The rate-limiting step in the transport of long-chain free fatty acids into the inner mitochondrial matrix is the one controlled by CPT1. The activity of CPT1 is regulated by malonyl-CoA, such that high intracellular levels of malonyl-CoA allosterically inhibit the activity of CPT1 (Park and Cook, 1998; Saggerson et al, 1992). In fact, high sensitivity of fetal CPT1 to inhibition by malonyl-CoA prevents the entry of free fatty acids into fetal mitochondria, and thus long-chain fatty acid oxidation probably does not occur to any significant level in the fetus (Prip-Buus et al, 1990; Saggerson and Carpenter, 1982). Malonyl-CoA is the product of the first committed step in the synthesis of fatty acids that is regulated by the biotin-requiring enzyme acetyl-CoA carboxylase:
CH3-CO-CoA + HCO3− +ATP → CO2H-CH2-CO-CoA + ADP + Pi
Fluctuations in tissue malonyl-CoA content parallel changes in acetyl-CoA carboxylase activity, which in turn is under the control of 5′-AMP–activated protein kinase; the CPT1/malonyl-CoA system is part of a fuel-sensing gauge, turning off and on fatty acid oxidation depending on the tissue’s energy demand (Kerner and Hoppel, 2000).
Once inside the mitochondria, acyl-CoA undergoes a set of four reactions, each catalyzed by a different enzyme (Bennett et al, 2000) (Fig. 2). This set of four reactions is repeated until the fatty acid is broken down into 2-carbon moieties of acetyl-CoA (one 3-carbon moiety propionyl-CoA is also produced if the initial fatty acid is composed of an odd number of carbons). The first step is catalyzed by very long-chain acyl-CoA dehydrogenase (VLCAD) and involves dehydrogenation of the C2-C3 bond (positions α and β) and production of a 2,3-enoyl-CoA. One molecule of flavine adenine dinucleotide (FAD) is reduced in the process:

Mitochondrial β-oxidation of fatty acids. The enzymes long-chain 2,3-enoyl-coenzyme A (CoA) hydratase (LHYD), long-chain l-3-hydroxyacyl-CoA dehydrogenase (LCHAD), and long-chain 3-ketoacyl-CoA thiolase (LKAT) are components of membrane-bound mitochondrial trifunctional protein (MTP). The medium- and short-chain homologs of these enzymes are nonmembrane–bound discrete proteins. VLCAD = very long-chain acyl-CoA dehydrogenase.
R-CH2-CH2-CO-SCoA + FAD → R-CH=CH-CO-SCoA + FADH2
The second step is catalyzed by long-chain 2,3-enoyl-CoA hydratase (LHYD), which hydrates the 2,3-enoyl-CoA across the C2-C3 double bond, so that the hydroxyl group is at the third carbon (the β carbon). The species generated is a stereospecific L-3-hydroxyacyl-CoA:
R-CH=CH-CO-SCoA + H2O → R-CHOHCH2- CO-SCoA
In the third step, catalyzed by LCHAD, the 3-hydroxy (β-hydroxy) position is oxidized to yield a 3-ketoacyl-CoA. This is accompanied by the reduction of one molecule of nicotinamide adenine dinucleotide (NAD+):
R-CHOHCH2-CO-SCoA + NAD+ → R-COCH2-CO-SCoA + NADH + H+
The last step involves thiolytic cleavage of the 3-ketoacyl-CoA to acetyl-CoA and an acyl-CoA that is shorter by two carbons. This step is catalyzed by the enzyme long-chain 3-ketoacyl-CoA thiolase (LKAT) and uses up a coenzyme A molecule:
R-COCH2-CO-SCoA + CoASH → R-CO-SCoA + CH3-CO-SCoA
The new acyl-CoA (shorter by two carbon atoms) re-enters the four-step β-oxidation cycle, and the process repeats itself until there is complete breakdown of the fatty acid into two-carbon moieties (and a single three-carbon moiety if the fatty acid was composed of an odd number of carbons).
As the chain-length of acyl-CoA decreases with each thiolytic cleavage, the above-mentioned enzymes specific for long-chain fatty acids lose their affinity. However, other similar enzymes exist in the mitochondrial matrix that have substrate specificities for fatty acids of shorter chain-lengths. These nonmembrane-bound enzymes include the following: long-chain acyl-CoA dehydrogenase (LCAD), medium-chain acyl-CoA dehydrogenase (MCAD), and short-chain acyl-CoA dehydrogenase (SCAD), which are homologs of each other and have catalytic activities similar to VLCAD; a short-chain 2,3-enoyl-CoA hydratase (SHYD), which is a homolog of LHYD; a medium/short-chain L-3-hydroxyacyl-CoA dehydrogenase (M/SCHAD), which is a homolog of LCHAD; and a medium-chain 3-ketoacyl-CoA thiolase (MKAT) and a short-chain 3-ketoacyl-CoA thiolase (SKAT), which are homologs of LKAT (Bennett et al, 2000; Wanders et al, 1999).
The complete β-oxidation reaction can be written as:
n FAD + n NAD+ + n CoASH + n H2O + H(CH2CH2)nCH2CO-SCoA
→
n FADH2 + n NADH + n H+ + (n+1) CH3CO-SCoA
This complex process is adequate for completely saturated fatty acids (eg, palmitic acid). For unsaturated fatty acids (eg, linoleic acid), two additional steps are required, catalyzed by 2,4-dienoyl-CoA reductase and enoyl-CoA isomerase, which allow for complete oxidation of the unsaturated fatty acids.
The acetyl-CoA generated via β-oxidation is directly utilized as an energy substrate within the citric acid cycle (skeletal muscle) or is channeled into ketogenesis or gluconeogenesis (liver). The propionyl-CoA is converted by a series of reactions into succinyl-CoA, which is utilized in the citric acid cycle. The reduced FAD and NAD+ are recycled after they lose their electrons to intermediates in the electron transport chain for generation of ATP.
Mitochondrial Trifunctional Protein (MTP)
The four enzymes involved in mitochondrial β-oxidation of long-chain fatty acids are actually just two proteins, both bound to the inner mitochondrial membrane. While VLCAD is one protein, the other three enzymes—LHYD, LCHAD, LKAT—are enzyme activities present in a single protein called the MTP (Carpenter et al, 1992; Uchida et al, 1992). The MTP is an hetero-octamer composed of four α and four β subunits; the α subunit harbors the long-chain 3-enoyl-CoA hydratase and LCHAD activities, whereas the β subunit is responsible for the long-chain 3-ketoacyl-CoA thiolase activity (Kamijo et al, 1993). The two subunits of MTP are encoded by separate genes, HADHA and HADHB, respectively, which are located contiguously on chromosome 2p23 (IJlst et al, 1996; Kamijo et al, 1994; Yang et al, 1996). A schematic representation of the MTP subunit cDNAs is shown in Figure 3.

Schematic representation of the structures of MTP subunit cDNAs.
Molecular Basis of LCHAD and MTP Deficiencies
MTP deficiency can be broadly classified into two biochemical and molecular patterns: one in which there exists a deficiency of LCHAD activity alone and another with deficiencies of all three enzyme activities (IJlst et al, 1996; Ushikubo et al, 1996). Of the two, isolated LCHAD deficiency seems to be the more common disorder.
The first molecular defect to be described for LCHAD deficiency was a G1528C mutation in exon 15 of the HADHA gene (IJlst et al, 1994; Sims et al, 1995), which remains the most common molecular basis for LCHAD deficiency (Ibdah et al, 1999; IJlst et al, 1995, 1996; Tyni et al, 1997a, 1998a; Wanders et al, 1999). This mutation alters amino acid 474 from glutamic acid to glutamine (E474Q), replacing the acidic and negatively charged side chain with a neutral, amide-containing residue. This affects the NAD+-binding site of LCHAD, leading to a loss of enzyme activity of LCHAD alone without affecting the other two enzymatic activities of MTP (Barycki et al, 1999). Other mutations described in patients with isolated LCHAD deficiency are rare (Ibdah et al, 1999).
The less common pattern of MTP deficiency involving a decrease in all three enzyme activities can be caused by mutations affecting either the HADHA or the HADHB genes. The HADHA gene defects include the following: a 71-bp deletion at position 110–180 (Ushikubo et al, 1996); a T845A mutation that substitutes aspartic acid for valine at residue 246, a T914A mutation that substitutes asparagine for isoleucine at residue 269, and a C871T mutation that creates a premature termination at residue 255 (Ibdah et al, 1998); mutations in the 5′ donor splice site following exon 3—a G to A substitution at the invariant position +1 and an A to G substitution at position +3, both apparently causing exon 3 skipping (Brackett et al, 1995); and a C to T mutation (C1678T) in exon 16 that creates a premature termination codon (R524Stop) in the LCHAD domain (Isaacs et al, 1996).
The HADHB gene mutations include the following: A788G, G182A, and G740A substitutions (Ushikubo et al, 1996); an exonic single T insertion at nucleotide position 777 that creates a new cryptic 5′ splice site leading to a 36-bp deletion at position 776–811 (Orii et al, 1997); and a G1331A transition (Orii et al, 1997).
The mutations in the HADHA and HADHB genes leading to a complete MTP deficiency either create truncated proteins or affect the stability of the interaction between the α and the β subunits of the hetero-octamer. The existence of both normal α and β subunits, and possibly their association, are important for stabilizing MTP (Ushikubo et al, 1996).
Features of LCHAD/MTP Deficiency in Individuals with Two Defective Genes (Homozygotes or Double Heterozygotes)
Case Report
The patient, a girl, was born via spontaneous vaginal delivery to a 17-year-old, G2P2, Caucasian mother. The mother apparently had an uneventful pregnancy. She denied any ethanol or drug use during the pregnancy. The child’s weight at birth was 3 kg, and her height at birth was 48.3 cm.
At 2 months of age, she was admitted to another hospital for upper respiratory tract infection and hypothermia (body temperature of 90.6°F). During her stay in the hospital, she had several episodes of seizures that were controlled with phenobarbital. Magnetic resonance imaging of the brain revealed a right posterior cerebrovascular accident. During this time, she had an episode of lactic acidosis, and a metabolic evaluation showed the following: blood glucose 25 mg/dl, serum lactic acid 3.8 mm, cerebrospinal fluid lactic acid 2.3 mm (normal 0.6 to 2.2), serum total carnitine 5 μm (normal 47 ± 11), serum free carnitine 1 μm (normal 38 ± 11), serum acylcarnitine 4 μm (normal 9 ± 5), normal serum ammonia, nonspecific pattern of serum amino acid profile, and negative urine amino acid profile. She was started on carnitine supplementation that she tolerated well. Subsequently she had three episodes of ear infection in the next 2 to 3 months but no further metabolic problems.
She was seen at Children’s Medical Center, Dallas, Texas at 6 months of age. She was alert and playful with good head control. Her height was 65.1 cm (38th percentile), weight was 7.2 kg (48th percentile), and head circumference was 40.3 cm (10th percentile). A skin biopsy was obtained for fibroblast culture. Enzymatic studies (Table 1) on the cultured fibroblasts showed depression of palmitate and myristate oxidation (approximately 10% and 40% of control values, respectively) with depressed LCHAD activity (about 4 standard deviations below the control’s mean value). The activities of SCHAD and SKAT were normal. A urinary organic acid analysis revealed an abnormal pattern with large excretions of 3-hydroxy-sebacic, 3-hydroxy-dodecanedioic, and unsaturated 3-hydroxy-tetradecanedioic acids consistent with the enzymatic observation of reduced LCHAD activity. A molecular genetic analysis revealed the child to be double heterozygote with two mutations in the α subunit of MTP (HADHA gene): G1528C and delT1967, the latter being a frame shift mutation that introduces a stop codon.
The patient started walking at about 1 year of age. When seen at 16 months of age she had good balance/walking coordination and was saying 6 to 8 recognizable single words; she had apparent normal developmental progression with her peers, normal muscle tone of 4/5, and normal muscle strength of 4.5/5. Her height and head circumference, however, continued to be less for her age and were 14th percentile and 3rd percentile, respectively, at 19 months of age. Her diet was modified to include medium-chain triglyceride oil for supplementation to achieve calories required for normal growth/development. Her last documented visit at Children’s Medical Center was at 25 months of age, at which time she was well. Shortly after this visit she was reported to have died suddenly and unexpectedly. Postmortem examination was not undertaken.
The child’s elder sibling, her half-brother, had no apparent medical problems. The family history was significant for malignancies in both paternal and maternal relatives.
Pathophysiology
Individuals with only one mutation in the genes for MTP (heterozygous) do not come to clinical attention because one normal gene apparently produces enough enzyme activity for catabolizing fatty acids, even in times of metabolic stress. The only exception is certain pregnancies as discussed later.
The patients with homozygous or double heterozygous defects in MTP can have varied clinical manifestations. Most of the cases come to clinical attention early in childhood, generally before the age of 3 years (Tyni and Pihko, 1999), with the symptoms being precipitated by intercurrent infections and associated decreased food intake. The subsequent decrease in blood glucose levels increases blood levels of catabolic hormones like glucagon and epinephrine, while the insulin levels fall. This causes mobilization of fatty acids, which are directed towards mitochondrial oxidation so that (a) NADH and FADH2 can be produced that can enter the mitochondrial electron transport chain to produce ATP and (b) acetyl-CoA can be produced for entry into the citric acid cycle, ketogenesis, and/or gluconeogenesis. Because the patients with reduced levels of long-chain fatty acid oxidation enzymes cannot oxidize fatty acids, the above goals cannot be achieved. This results in hypoglycemia that is characteristically associated with low blood ketone levels or hypoketotic hypoglycemia. This is seen with many fatty acid oxidation disorders, especially those affecting the long-chain fatty acids, and is not unique to LCHAD/MTP deficiencies. Besides hypoketotic hypoglycemia, other relatively nonspecific biochemical findings in acutely ill patients include elevated serum aminotransferases, elevated serum creatine kinase, lactic acidosis, and hyperammonemia. More diagnostic are elevated levels of long-chain 3-hydroxyfatty acids, 3-hydroxyacylcarnitines, 3-hydroxyacyl-CoAs, and 3-hydroxydicarboxylic acids in serum and urine. These compounds represent metabolite build-up proximal to the enzyme defect. As a result of the formation of hydroxyacylcarnitines, blood carnitine levels also fall.
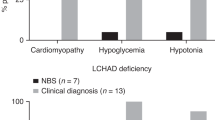
Not only are these biochemical changes of diagnostic importance, they contribute to the pathology seen in these patients. Because of decreased lipid breakdown, there is an abnormally increased amount of fat in various organs, including liver, skeletal muscle, myocardium, renal tubules, pancreas, and lungs (Duran et al, 1991; Tyni at al, 1997b). Also, the accumulated intermediates of long-chain fatty acid oxidation are toxic to biomembranes and macromolecules as a result of their detergent-like properties (Wojtczak and Schonfeld, 1993). They can damage mitochondria and the respiratory enzymes, causing a secondary reduction in respiratory chain function (Das et al, 2000; Tyni et al, 1996). An increase in size and number of mitochondria, as well as mitochondrial damage in the form of swelling, and irregular cristae has been demonstrated using electron microscopy (Hagenfeldt et al, 1995; Rocchiccioli et al, 1990; Tyni et al, 1996, 1997b). The infants also manifest evidence of cardiomyopathy with dilated cardiomegaly and poor heart sounds, hypotonia, hepatomegaly, Reye syndrome–like encephalopathy, growth retardation, and failure to thrive. Some patients first manifest in late childhood and even early adulthood with clinical features of skeletal myopathy. They have exercise-induced muscle pain, rhabdomyolysis, and myoglobinuria. The patients who survive longer may develop peripheral neuropathy and pigmentary retinopathy. Other less common features include microcephaly, cholestatic liver disease, massive hepatic necrosis, anemia, thrombocytopenia, and hypoparathyroidism. There may be a history of sudden death of a sibling or a history of maternal illness during pregnancy (Amirkhan et al, 1997; Bertini et al, 1992; Dionisi-Vici et al, 1991, 1996; Hagenfeldt et al, 1995; Lawlor and Kalina, 1997; Martins et al, 1996; Miyajima et al, 1997; Pons et al, 1996; Przyrembel et al, 1991; Ribes et al, 1992; Rocchiccioli et al, 1990; Schaefer et al, 1996; Schrijver-Wieling et al, 1997; Sewell et al, 1994; Tyni et al, 1997a, 1997c, 1998b, 1998c; Wanders et al, 1990). The pathologic findings in the brain include vacuoles in the cerebellar dentate nucleus, lateral geniculate nucleus, pontine nuclei, and deep cerebral gray matter (Tyni et al, 1997b). The ophthalmic pathology is characterized by progressive atrophy of the retinal pigmentary epithelium and choroids, which gives a mottled appearance to the fundus. There is relative central macular sparing (Schrijver-Wieling et al, 1997; Tyni at al, 1998b, 1998c).
Pregnancy and Fatty Acid Oxidation Defects
The etiology of a number of important diseases unique to pregnancy, including preeclampsia, HELLP syndrome, hyperemesis gravidarum, AFLP, and maternal floor infarct of the placenta is not clear, although hypotheses abound in the literature (Bacq, 1998; Dietl, 2000; Eliakim et al, 2000; Rath et al, 2000; Vernof et al, 1992). These diseases have recently been shown in some cases to be associated with LCHAD deficiency in the fetus (Ibdah et al, 1999; Isaacs et al, 1996; Matern et al, 2001b; Pollitt, 1995; Sims et al, 1995; Treem et al, 1994; Tyni et al, 1998a; Wilcken et al, 1993). Tyni et al (1998a) reported that preeclampsia, HELLP syndrome, and AFLP occurred in 31% and intrahepatic cholestasis in 10% of pregnancies with a LCHAD-deficient fetus but in none of the pregnancies at risk for LCHAD deficiency with a healthy fetus. This apparent association of fetal LCHAD deficiency with maternal disease has led to the discovery of other similar associations. Innes et al (2000) identified absent CPT1 activity in two siblings whose mother had AFLP and hyperemesis gravidarum in both pregnancies. Matern et al (2001a) detected SCAD deficiency in an infant evaluated because his mother had AFLP, and Nelson et al (2000) reported a pregnancy complicated by severe preeclampsia and HELLP syndrome in which the fetus was subsequently shown to have MCAD deficiency.
However, the mechanism whereby a fatty acid oxidation defect in the fetus causes overt maternal disease is yet to be clearly elucidated. The heterozygous mother is not symptomatic until she becomes pregnant with a fetus who is homozygous for the defect. The accumulation of potentially toxic intermediate products of fatty acid metabolism in the mother can theoretically occur from three sources: the heterozygous mother herself, the homozygous fetus, or the homozygous placenta, which has the same genetic makeup as the fetus. The mother seems an unlikely source because this would imply that HELLP syndrome and AFLP should occur in metabolically stressed nonpregnant female and male heterozygotes. Only a single example of an infant heterozygous for LCHAD deficiency and progressive liver disease has been reported. This infant had concurrent HIV infection, so that it was difficult to define the etiology of the liver disease, although the introduction of a low-fat diet resulted in a marked improvement in the child’s clinical status. However, the molecular basis for LCHAD deficiency was not defined (Hicks et al, 1995). The homozygous fetus is unlikely to produce intermediates of fatty acid oxidation because glucose is the main energy source for the fetus, and fetal fatty acid oxidation is low (Herrera and Amusquivar, 2000). The low levels of CPT I in the fetus, as well as high sensitivity of fetal CPT I to inhibition by malonyl-CoA, prevent the entry of free fatty acids into the fetal mitochondria, where the β-oxidation of fatty acids occurs (Prip-Buus et al, 1990; Saggerson and Carpenter, 1982). We recently showed that placenta expresses the active enzymes of fatty acid oxidation (Rakheja et al, 2002; Shekhawat et al, 2001), including CPT1 (our unpublished data), which is down-regulated in the fetus, and a defective placenta seems like a good candidate for initiating the chain of events leading to symptoms in the mother.
How the accumulated intermediates of fatty acid oxidation translate into maternal diseases is yet to be demonstrated. Hypothetically, these intermediates may act as free radicals causing damage to cell membranes and organelles. In LCHAD deficiency, the accumulated metabolic intermediates include long-chain 3-hydroxy-fatty acids, 3-hydroxyacylcarnitines, 3-hydroxyacyl-CoAs, and 3-hydroxy-dicarboxylic acids, which in high concentrations can injure cell membranes, potentiate free radical-induced lipid peroxidation, inhibit Na+-K+-ATPase, uncouple mitochondrial oxidative phosphorylation, and damage mitochondria (Kramer and Weglicki, 1985; Mak et al, 1986; Singh et al, 1989; Wojtczak and Schonfeld, 1993). Widespread damage to the maternal endothelium may cause the release of inflammatory mediators, leading to a systemic illness with multiple organ damage. In fact, damage to vascular endothelium may be an early event in the pathophysiology of preeclampsia (Gratacos, 2000; Roberts and Cooper, 2001) and oxidative stress is favored as the cause for endothelial damage in preeclampsia (Roberts and Hubel, 1999). The origin of this oxidative stress may be in the placenta. It has been suggested that the placenta may be an important source of lipid peroxides in preeclampsia (Gratacos, 2000). Wang and Walsh (1998) concluded from their study that placental mitochondria contribute to the abnormal increase in lipid peroxidation that occurs in preeclamptic placentas by both an increase in the mitochondrial number and an increase in their susceptibility to lipid peroxidation. Not only is the mitochondrial number increased in trophoblastic cells from preeclamptic placentas, but the trophoblastic mitochondria also show swelling and a loss of cristae (Shanklin and Sibai, 1989), a change that has also been detected in mitochondria from maternal tissues in the setting of preeclampsia (Shanklin and Sibai, 1990). Interestingly, an increase in mitochondrial number, mitochondrial swelling, and irregular mitochondrial cristae have been seen in the skeletal muscle cells of four of seven children with LCHAD deficiency (Tyni et al, 1996). Similarly, a decrease in the activity of respiratory chain complexes has been described in trophoblastic cells from preeclamptic placentas (Matsubara et al, 1997) as well as in skeletal muscle cells of children with LCHAD deficiency (Tyni et al, 1996). It is thus not difficult to hypothesize a possible mechanism for development of preeclampsia in mothers carrying an LCHAD-deficient fetus (Fig. 4). A similar argument could be presented for other mitochondrial disorders in which there is mitochondrial damage. However, there must be other factors still to be determined that influence the fetal-placental-maternal environment, because some heterozygous mothers carrying a homozygous LCHAD-deficient fetus do not develop pregnancy-related disease (Tyni et al, 1998a).

A hypothetical schematic representation illustrating the cause of preeclampsia in a mother carrying a fetus/placenta with mitochondrial fatty acid oxidation defect.
Diagnosis
The diagnosis of fatty acid oxidation disorders, or for that matter any other error of metabolism, can be based on metabolite analysis, enzyme activity measurement, or molecular analysis. While fibroblasts from skin biopsy samples are easily obtained for postnatal diagnosis, chorionic villi, amniocytes, or even fetal cells in maternal circulation are suitable samples for prenatal analysis (Jauniaux et al, 2000; Jenkins and Wapner, 1999; Pertl and Bianchi, 1999).
Metabolite analysis is based on recognizing the different patterns of metabolite accumulation specific to different fatty acid oxidation enzyme defects. In LCHAD deficiency there is a predominant increase in long-chain 3-hydroxy-acyl derivatives, which can be measured by gas chromatography-mass spectrometry or tandem mass spectrometry (Jones et al, 2000; Roe and Roe, 1999). Shen et al (2000) measured acylcarnitines in cells and media of cultured fibroblasts of patients with LCHAD deficiency. After incubation with palmitate (a 16:0 fatty acid), LCHAD-deficient fibroblasts showed elevation of hydroxypalmitoyl- and palmitoyl-carnitine and reduction of C10- and shorter acylcarnitines. Similarly, after incubation with linoleate (an 18:2 fatty acid), there was an increase in C14:2-, C18:2-, and hydroxy-C18:2- acylcarnitines and a reduction in C10:1-acylcarnitines. Incubation with decanoate and octanoate (medium-chain fatty acids) failed to show accumulation of hydroxylated acylcarnitines. A different metabolite accumulation profile would point to a different specific enzyme defect, and such in vitro studies also can be performed on amniocytes (Nada et al, 1996; Roe and Roe, 1999).
Conventional enzyme assays have been described for 3-hydroxyacyl-CoA dehydrogenases (Bennett et al, 1996; Venizelos et al, 1994; Wanders et al, 1990) and have been used for prenatal diagnosis of LCHAD deficiency (Perez-Cerda et al, 1993). All enzymes of mitochondrial fatty acid oxidation are readily expressed in chorionic villi biopsy specimens as well as in cultured chorionic villous fibroblasts and amniocytes (Wanders et al, 1999).
Molecular identification of specific genetic mutations is gaining increasing use for prenatal as well as postnatal diagnosis of inborn errors of metabolism (Evans and Levy, 1999). Ibdah et al (2001) demonstrated the feasibility of performing rapid molecular prenatal diagnosis on chorionic villi and amniocytes in families with known MTP defects. However, metabolite and enzyme measurements will continue to have a central diagnostic role in screening at least until methods to detect all possible mutations become available, a difficult if not an improbable task at the present time.
A definitive prenatal diagnosis affords a choice of terminating a pregnancy, as well as preparing the obstetrician and the neonatologist for upcoming problems if the pregnancy is carried to term. An emerging alternative strategy that avoids termination of pregnancy is preimplantation diagnosis (Wells and Sherlock, 1998) using cells from in vitro fertilization–generated embryos. This technique has recently been applied for diagnosis of MCAD deficiency (Ioulianos et al, 2000; Sermon et al, 2000).
Management
The basic management goal is to prevent or minimize long-chain fatty acid oxidation. This is accomplished in an acutely ill patient by rapid intravenous infusion of 10% glucose solutions. The secondary carnitine deficiency may be corrected by carnitine administration. The long-term management is dietary, with avoidance of fasting and a diet low in long-chain fatty acids (providing mainly the essential fatty acids) and compensated by medium-chain fatty acids (Gillingham et al, 1999; Saudubray et al, 1999). Docosahexaenoic acid, an ω-3 fatty acid, is being tried as a dietary supplement to treat the retinopathy (Harding et al, 1999).
References
Amirkhan RH, Timmons CF, Brown KO, Weinberger MJ, and Bennett MJ (1997). Clinical, biochemical, and morphologic investigations of a case of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Arch Pathol Lab Med 121: 730–734.
Bacq Y (1998). Acute fatty liver of pregnancy. Semin Perinatol 22: 134–140.
Barycki JJ, O'Brien LK, Bratt JM, Zhang R, Sanishvili R, Strauss AW, and Banaszak LJ (1999). Biochemical characterization and crystal structure determination of human heart short chain L-3-hydroxyacyl-CoA dehydrogenase provide insights into catalytic mechanism. Biochemistry 38: 5786–5798.
Bennett MJ, Rinaldo P, and Strauss AW (2000). Inborn errors of mitochondrial fatty acid oxidation. Crit Rev Clin Lab Sci 37: 1–44.
Bennett MJ, Weinberger MJ, Kobori JA, Rinaldo P, and Burlina AB (1996). Mitochondrial short-chain L-3-hydroxyacyl-coenzyme A dehydrogenase deficiency: A new defect of fatty acid oxidation. Pediatr Res 39: 185–188.
Bertini E, Dionisi-Vici C, Garavaglia B, Burlina AB, Sabatelli M, Rimoldi M, Bartuli A, Sabetta G, and DiDonato S (1992). Peripheral sensory-motor polyneuropathy, pigmentary retinopathy, and fatal cardiomyopathy in long-chain 3-hydroxy-acyl-CoA dehydrogenase deficiency. Eur J Pediatr 151: 121–126.
Brackett JC, Sims HF, Rinaldo P, Shapiro S, Powell CK, Bennett MJ, and Strauss AW (1995). Two alpha subunit donor splice site mutations cause human trifunctional protein deficiency. J Clin Invest 95: 2076–2082.
Carpenter K, Pollitt RJ, and Middleton B (1992). Human liver long-chain 3-hydroxyacyl-coenzyme A dehydrogenase is a multifunctional membrane-bound beta-oxidation enzyme of mitochondria. Biochem Biophys Res Commun 183: 443–448.
Das AM, Fingerhut R, Wanders RJ, and Ullrich K (2000). Secondary respiratory chain defect in a boy with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Possible diagnostic pitfalls. Eur J Pediatr 159: 243–246.
Dietl J (2000). The pathogenesis of pre-eclampsia: New aspects. J Perinat Med 28: 464–471.
Dionisi-Vici C, Burlina AB, Bertini E, Bachmann C, Mazziotta MR, Zacchello F, Sabetta G, and Hale DE (1991). Progressive neuropathy and recurrent myoglobinuria in a child with long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. J Pediatr 118: 744–746.
Dionisi-Vici C, Garavaglia B, Burlina AB, Bertini E, Saponara I, Sabetta G, and Taroni F (1996). Hypoparathyroidism in mitochondrial trifunctional protein deficiency. J Pediatr 129: 159–162.
Duran M, Wanders RJ, de Jager JP, Dorland L, Bruinvis L, Ketting D, IJlst L, and van Sprang FJ (1991). 3-Hydroxydicarboxylic aciduria due to long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency associated with sudden neonatal death: Protective effect of medium-chain triglyceride treatment. Eur J Pediatr 150: 190–195.
Eliakim R, Abulafia O, and Sherer DM (2000). Hyperemesis gravidarum: A current review. Am J Perinatol 17: 207–218.
Evans MI and Levy HL (1999). The future of newborn screening belongs to obstetricians. Acta Paediatr Suppl 88: 55–57.
Gillingham M, Van Calcar S, Ney D, Wolff J, and Harding C (1999). Dietary management of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD): A case report and survey. J Inherit Metab Dis 22: 123–131.
Gratacos E (2000). Lipid-mediated endothelial dysfunction: A common factor to preeclampsia and chronic vascular disease. Eur J Obstet Gynecol Reprod Biol 92: 63–66.
Harding CO, Gillingham MB, van Calcar SC, Wolff JA, Verhoeve JN, and Mills MD (1999). Docosahexaenoic acid and retinal function in children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 22: 276–280.
Hagenfeldt L, Venizelos N, and von Dobeln U (1995). Clinical and biochemical presentation of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 18: 245–248.
Herrera E and Amusquivar E (2000). Lipid metabolism in the fetus and the newborn. Diabetes Metab Res Rev 16: 202–210.
Hicks P, Bennett MJ, Squires J, and Ramilo O (1995). Heterozygosity for long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency and deterioration in liver function in a newborn infant infected with human immunodeficiency virus. J Pediatr 127: 599–602.
Ibdah JA, Bennett MJ, Rinaldo P, Zhao Y, Gibson B, Sims HF, and Strauss AW (1999). A fetal fatty-acid oxidation disorder as a cause of liver disease in pregnant women. N Engl J Med 340: 1723–1731.
Ibdah JA, Tein I, Dionisi-Vici C, Bennett MJ, IJlst L, Gibson B, Wanders RJ, and Strauss AW (1998). Mild trifunctional protein deficiency is associated with progressive neuropathy and myopathy and suggests a novel genotype-phenotype correlation. J Clin Invest 102: 1193–1199.
Ibdah JA, Zhao Y, Viola J, Gibson B, Bennett MJ, and Strauss AW (2001). Molecular prenatal diagnosis in families with fetal mitochondrial trifunctional protein mutations. J Pediatr 138: 396–399.
IJlst L, Ruiter JP, Hoovers JM, Jakobs ME, and Wanders RJ (1996). Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein alpha subunit gene. J Clin Invest 98: 1028–1033.
IJlst L, Uskikubo S, Kamijo T, Hashimoto T, Ruiter JP, de Klerk JB, and Wanders RJ (1995). Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: High frequency of the G1528C mutation with no apparent correlation with the clinical phenotype. J Inherit Metab Dis 18: 241–244.
IJlst L, Wanders RJ, Ushikubo S, Kamijo T, and Hashimoto T (1994). Molecular basis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Identification of the major disease-causing mutation in the alpha-subunit of the mitochondrial trifunctional protein. Biochim Biophys Acta 1215: 347–350.
Innes AM, Seargeant LE, Balachandra K, Roe CR, Wanders RJ, Ruiter JP, Casiro O, Grewar DA, and Greenberg CR (2000). Hepatic carnitine palmitoyl transferase I deficiency presenting as maternal illness in pregnancy. Pediatr Res 47: 43–45.
Ioulianos A, Wells D, Harper JC, and Delhanty JD (2000). A successful strategy for preimplantation diagnosis of medium-chain acyl-CoA dehydrogenase (MCAD) deficiency. Prenat Diagn 20: 593–598.
Isaacs JD Jr, Sims HF, Powell CK, Bennett MJ, Hale DE, Treem WR, and Strauss AW (1996). Maternal acute fatty liver of pregnancy associated with fetal trifunctional protein deficiency: Molecular characterization of a novel maternal mutant allele. Pediatr Res 40: 393–398.
Jauniaux E, Pahal GS, and Rodeck CH (2000). What invasive procedure to use in early pregnancy? Baillieres Best Pract Res Clin Obstet Gynaecol 14: 651–662.
Jenkins TM and Wapner RJ (1999). First trimester prenatal diagnosis: Chorionic villus sampling. Semin Perinatol 23: 403–413.
Jones PM, Quinn R, Fennessey PV, Tjoa S, Goodman SI, Fiore S, Burlina AB, Rinaldo P, Boriack RL, and Bennett MJ (2000). Improved stable isotope dilution-gas chromatography-mass spectrometry method for serum or plasma free 3-hydroxy-fatty acids and its utility for the study of disorders of mitochondrial fatty acid beta-oxidation. Clin Chem 46: 149–155.
Kamijo T, Aoyama T, Komiyama A, and Hashimoto T (1994). Structural analysis of cDNAs for subunits of human mitochondrial fatty acid beta-oxidation trifunctional protein. Biochem Biophys Res Commun 199: 818–825.
Kamijo T, Aoyama T, Miyazaki J, and Hashimoto T (1993). Molecular cloning of the cDNAs for the subunits of rat mitochondrial fatty acid beta-oxidation multienzyme complex: Structural and functional relationships to other mitochondrial and peroxisomal beta-oxidation enzymes. J Biol Chem 268: 26452–26460.
Kerner J and Hoppel C (2000). Fatty acid import into mitochondria. Biochim Biophys Acta 1486: 1–17.
Kramer JH and Weglicki WB (1985). Inhibition of sarcolemmal Na+-K+-ATPase by palmitoyl carnitine: Potentiation by propranolol. Am J Physiol 248: H75–H81.
Lawlor DP and Kalina RE (1997). Pigmentary retinopathy in long chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Am J Ophthalmol 123: 846–848.
Mak IT, Kramer JH, and Weglicki WB (1986). Potentiation of free radical-induced lipid peroxidative injury to sarcolemmal membranes by lipid amphiphiles. J Biol Chem 261: 1153–1157.
Martins E, Costa A, Silva E, Medina M, Cardoso ML, Vianey-Saban C, Divry P, and Vilarinho L (1996). Lethal dilated cardiomyopathy due to long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 19: 373–374.
Matern D, Hart P, Murtha AP, Vockley J, Gregersen N, Millington DS, and Treem WR (2001a). Acute fatty liver of pregnancy associated with short-chain acyl-coenzyme A dehydrogenase deficiency. J Pediatr 138: 585–588.
Matern D, Shehata BM, Shekhawat P, Strauss AW, Bennett MJ, and Rinaldo P (2001b). Placental floor infarct complicating the pregnancy of a fetus with long chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Mol Genet Metab 72: 265–268.
Matsubara S, Minakami H, Sato I, and Saito T (1997). Decrease in cytochrome c oxidase activity detected cytochemically in the placental trophoblast of patients with pre-eclampsia. Placenta 18: 255–259.
McGarry JD and Brown NF (1997). The mitochondrial carnitine palmitoyl transferase system: From concept to molecular analysis. Eur J Biochem 244: 1–14.
Miyajima H, Orii KE, Shindo Y, Hashimoto T, Shinka T, Kuhara T, Matsumoto I, Shimizu H, and Kaneko E (1997). Mitochondrial trifunctional protein deficiency associated with recurrent myoglobinuria in adolescence. Neurology 49: 833–837.
Nada MA, Vianey-Saban C, Roe CR, Ding JH, Mathieu M, Wappner RS, Bialer MG, McGlynn JA, and Mandon G (1996). Prenatal diagnosis of mitochondrial fatty acid oxidation defects. Prenat Diagn 16: 117–124.
Nelson J, Lewis B, and Walters B (2000). The HELLP syndrome associated with fetal medium-chain acyl-CoA dehydrogenase deficiency. Inherit Metab Dis 23: 518–519.
Orii KE, Aoyama T, Wakui K, Fukushima Y, Miyajima H, Yamaguchi S, Orii T, Kondo N, and Hashimoto T (1997). Genomic and mutational analysis of the mitochondrial trifunctional protein beta-subunit (HADHB) gene in patients with trifunctional protein deficiency. Hum Mol Genet 6: 1215–1224.
Park EA and Cook GA (1998). Differential regulation in the heart of mitochondrial carnitine palmitoyltransferase-I muscle and liver isoforms. Mol Cell Biochem 180: 27–32.
Perez-Cerda C, Merinero B, Jimenez A, Garcia MJ, Sanz P, IJlst L, Wanders RJ, and Ugarte M (1993). First report of prenatal diagnosis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency in a pregnancy at risk. Prenat Diagn 13: 529–533.
Pertl B and Bianchi DW (1999). First trimester prenatal diagnosis: Fetal cells in the maternal circulation. Semin Perinatol 23: 393–402.
Pollitt RJ (1995). Disorders of mitochondrial long-chain fatty acid oxidation. J Inherit Metab Dis 18: 473–490.
Pons R, Roig M, Riudor E, Ribes A, Briones P, Ortigosa L, Baldellou A, Gil-Gibernau J, Olesti M, Navarro C, and Wanders RJ (1996). The clinical spectrum of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Pediatr Neurol 14: 236–243.
Prip-Buus C, Pegorier JP, Duee PH, Kohl C, and Girard J (1990). Evidence that the sensitivity of carnitine palmitoyl transferase I to inhibition by malonyl-CoA is an important site of regulation of hepatic fatty acid oxidation in the fetal and newborn rabbit: Perinatal development and effects of pancreatic hormones in cultured rabbit hepatocytes. Biochem J 269: 409–415.
Przyrembel H, Jakobs C, IJlst L, de Klerk JB, and Wanders RJ (1991). Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 14: 674–680.
Rakheja D, Bennett MJ, Foster BM, Domiati-Saad R, and Rogers BB (In press, 2002). Evidence for fatty acid oxidation in human placenta, and the relationship of fatty acid oxidation enzyme activities with gestational age. Placenta.
Rath W, Faridi A, and Dudenhausen JW (2000). HELLP syndrome. J Perinat Med 28: 249–260.
Ribes A, Riudor E, Navarro C, Boronat M, Marti M, and Hale DE (1992). Fatal outcome in a patient with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 15: 278–279.
Rifai N, Bachorik PS, and Albers JJ (2001). Lipids, lipoproteins, and apolipoproteins. In: Burtis CA, and Ashwood ER, editors. Tietz fundamentals of clinical chemistry. Philadelphia: W B Saunders, 468.
Roberts JM and Cooper DW (2001). Pathogenesis and genetics of pre-eclampsia. Lancet 357: 53–56.
Roberts JM and Hubel CA (1999). Is oxidative stress the link in the two-stage model of pre-eclampsia? Lancet 354: 788–789.
Rocchiccioli F, Wanders RJ, Aubourg P, Vianey-Liaud C, IJlst L, Fabre M, Cartier N, and Bougneres PF (1990). Deficiency of long-chain 3-hydroxyacyl-CoA dehydrogenase: A cause of lethal myopathy and cardiomyopathy in early childhood. Pediatr Res 28: 657–662.
Roe CR and Roe DS (1999). Recent developments in the investigation of inherited metabolic disorders using cultured human cells. Mol Genet Metab 68: 243–257.
Saggerson ED and Carpenter CA (1982). Regulation of hepatic carnitine palmitoyl transferase activity during the fetal-neonatal transition. FEBS Lett 150: 177–180.
Saggerson D, Ghadiminejad I, and Awan M (1992). Regulation of mitochondrial carnitine palmitoyl transferases from liver and extrahepatic tissues. Adv Enzyme Regul 32: 285–306.
Saudubray JM, Martin D, de Lonlay P, Touati G, Poggi-Travert F, Bonnet D, Jouvet P, Boutron M, Slama A, Vianey-Saban C, Bonnefont JP, Rabier D, Kamoun P, and Brivet M (1999). Recognition and management of fatty acid oxidation defects: A series of 107 patients. J Inherit Metab Dis 22: 488–502.
Schaefer J, Jackson S, Dick DJ, and Turnbull DM (1996). Trifunctional enzyme deficiency: Adult presentation of a usually fatal beta-oxidation defect. Ann Neurol 40: 597–602.
Schoeman MN, Batey RG, and Wilcken B (1991). Recurrent acute fatty liver of pregnancy associated with a fatty-acid oxidation defect in the offspring. Gastroenterology 100: 544–548.
Schrijver-Wieling I, van Rens GH, Wittebol-Post D, Smeitink JA, de Jager JP, de Klerk HB, and van Lith GH (1997). Retinal dystrophy in long chain 3-hydroxy-acyl-coA dehydrogenase deficiency. Br J Ophthalmol 81: 291–294.
Sermon K, Henderix P, Lissens W, De Vos A, Vandervorst M, Vanderfaeillie A, Vamos E, Van Steirteghem A, and Liebaers I (2000). Preimplantation genetic diagnosis for medium-chain acyl-CoA dehydrogenase (MCAD) deficiency. Mol Hum Reprod 6: 1165–1168.
Sewell AC, Bender SW, Wirth S, Munterfering H, IJlst L, and Wanders RJ (1994). Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: A severe fatty acid oxidation disorder. Eur J Pediatr 153: 745–750.
Shanklin DR and Sibai BM (1989). Ultrastructural aspects of preeclampsia. I. Placental bed and uterine boundary vessels. Am J Obstet Gynecol 161: 735–741.
Shanklin DR and Sibai BM (1990). Ultrastructural aspects of preeclampsia. II. Mitochondrial changes. Am J Obstet Gynecol 163: 943–953.
Shekhawat PS, Bennett MJ, Rakheja D, and Strauss AW (2001). Fatty acid oxidation (FAO) in normal human placenta: Developmental expression and activity of enzymes of mitochondria beta oxidation (Abstract). Pediatr Res 49: 55.
Shen JJ, Matern D, Millington DS, Hillman S, Feezor MD, Bennett MJ, Qumsiyeh M, Kahler SG, Chen YT, and Van Hove JL (2000). Acylcarnitines in fibroblasts of patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency and other fatty acid oxidation disorders. J Inherit Metab Dis 23: 27–44.
Sims HF, Brackett JC, Powell CK, Treem WR, Hale DE, Bennett MJ, Gibson B, Shapiro S, and Strauss AW (1995). The molecular basis of pediatric long chain 3-hydroxyacyl-CoA dehydrogenase deficiency associated with maternal acute fatty liver of pregnancy. Proc Natl Acad Sci U S A 92: 841–845.
Singh AK, Yoshida Y, Garvin AJ, and Singh I (1989). Effect of fatty acids and their derivatives on mitochondrial structures. J Exp Pathol 4: 9–15.
Treem WR, Rinaldo P, Hale DE, Stanley CA, Millington DS, Hyams JS, Jackson S, and Turnbull DM (1994). Acute fatty liver of pregnancy and long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Hepatology 19: 339–345.
Tyni T, Ekholm E, and Pihko H (1998a). Pregnancy complications are frequent in long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Am J Obstet Gynecol 178: 603–608.
Tyni T, Pihko H, and Kivela T (1998b). Ophthalmic pathology in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation. Curr Eye Res 17: 551–559.
Tyni T, Kivela T, Lappi M, Summanen P, Nikoskelainen E, and Pihko H (1998c). Ophthalmologic findings in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation: A new type of hereditary metabolic chorioretinopathy. Ophthalmology 105: 810–824.
Tyni T, Majander A, Kalimo H, Rapola J, and Pihko H (1996). Pathology of skeletal muscle and impaired respiratory chain function in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency with the G1528C mutation. Neuromuscul Disord 6: 327–337.
Tyni T, Palotie A, Viinikka L, Valanne L, Salo MK, von Dobeln U, Jackson S, Wanders R, Venizelos N, and Pihko H (1997a). Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency with the G1528C mutation: Clinical presentation of thirteen patients. J Pediatr 130: 67–76.
Tyni T and Pihko H (1999). Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Acta Paediatr 88: 237–245.
Tyni T, Rapola J, Paetau A, Palotie A, and Pihko H (1997b). Pathology of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency caused by the G1528C mutation. Pediatr Pathol Lab Med 17: 427–447.
Tyni T, Rapola J, Palotie A, and Pihko H (1997c). Hypoparathyroidism in a patient with long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency caused by the G1528C mutation. J Pediatr 131: 766–768.
Uchida Y, Izai K, Orii T, and Hashimoto T (1992). Novel fatty acid beta-oxidation enzymes in rat liver mitochondria. II. Purification and properties of enoyl-coenzyme A (CoA) hydratase/3-hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase trifunctional protein. J Biol Chem 267: 1034–1041.
Ushikubo S, Aoyama T, Kamijo T, Wanders RJ, Rinaldo P, Vockley J, and Hashimoto T (1996). Molecular characterization of mitochondrial trifunctional protein deficiency: Formation of the enzyme complex is important for stabilization of both alpha- and beta-subunits. Am J Hum Genet 58: 979–988.
Veldee MS (2001). Nutritional assessment, therapy, and monitoring. In: Burtis CA, and Ashwood ER, editors. Tietz fundamentals of clinical chemistry. Philadelphia: W B Saunders, 941.
Venizelos N, IJlst L, Wanders RJ, and Hagenfeldt L (1994). Beta-oxidation enzymes in fibroblasts from patients with 3-hydroxydicarboxylic aciduria. Pediatr Res 36: 111–114.
Vernof KK, Benirschke K, Kephart GM, Wasmoen TL, and Gleich GJ (1992). Maternal floor infarction: Relationship to X cells, major basic protein, and adverse perinatal outcome. Am J Obstet Gynecol 167: 1355–1363.
Wanders RJ, Duran M, IJlst L, de Jager JP, van Gennip AH, Jakobs C, Dorland L, and van Sprang FJ (1989). Sudden infant death and long-chain 3-hydroxyacyl-CoA dehydrogenase. Lancet 2: 52–53.
Wanders RJ, IJlst L, van Gennip AH, Jakobs C, de Jager JP, Dorland L, van Sprang FJ, and Duran M (1990). Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Identification of a new inborn error of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis 13: 311–314.
Wanders RJ, Vreken P, den Boer ME, Wijburg FA, van Gennip AH, and IJlst L (1999). Disorders of mitochondrial fatty acyl-CoA beta-oxidation. J Inherit Metab Dis 22: 442–487.
Wang Y and Walsh SW (1998). Placental mitochondria as a source of oxidative stress in pre-eclampsia. Placenta 19: 581–586.
Wells D and Sherlock JK (1998). Strategies for preimplantation genetic diagnosis of single gene disorders by DNA amplification. Prenat Diagn 18: 1389–1401.
Wilcken B, Leung KC, Hammond J, Kamath R, and Leonard JV (1993). Pregnancy and fetal long-chain 3-hydroxyacyl coenzyme A dehydrogenase deficiency. Lancet 341: 407–408.
Wojtczak L and Schonfeld P (1993). Effect of fatty acids on energy coupling processes in mitochondria. Biochim Biophys Acta 1183: 41–57.
Yang B-Z, Heng HHQ, Ding J-H, and Roe CR (1996). The genes for the α and β subunits of the mitochondrial trifunctional protein are both located in the same region of the human chromosome 2p23. Genomics 37: 141–143.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rakheja, D., Bennett, M. & Rogers, B. Long-Chain L-3-Hydroxyacyl-Coenzyme A Dehydrogenase Deficiency: A Molecular and Biochemical Review. Lab Invest 82, 815–824 (2002). https://doi.org/10.1097/01.LAB.0000021175.50201.46
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.LAB.0000021175.50201.46
