
Abstract
Papillary thyroid microcarcinomas (measuring 1 cm or less in diameter) are very common thyroid tumors, which are present in 10% to 35% of post-mortem histopathological examinations of individuals whose death was due to a cause other than thyroid cancer. The molecular basis of this tumor is still poorly understood. Somatic mutations are better characterized in clinically evident papillary thyroid carcinomas (PTCs), the most common involving the proto-oncogene RET, which maps to 10q11.2. Molecular alterations of RET always lead to intra- or interchromosomal rearrangements. In this study we have investigated the status of RET in 21 microcarcinomas, by means of interphase fluorescence in situ hybridization (FISH). RET was rearranged in 52% of microcarcinomas, a statistically significant higher frequency than that found previously in clinically evident PTCs using the same technique. Moreover, interphase FISH allowed us to detect a putative novel type of rearrangement in a microcarcinoma, and we observed trisomies of chromosome 10 and other chromosomes in two adenomas surrounding two of the microcarcinomas. The strikingly high frequency of RET rearrangements in microcarcinomas strongly suggests that RET plays a role in the initiation of thyroid tumorigenesis but does not seem to be necessary for the further progression of the tumor.
Similar content being viewed by others


Introduction
Papillary thyroid carcinoma (PTC) represents approximately 70% of all thyroid carcinomas and includes numerous morphological variants (Parkin et al, 1999). Tumors measuring 1 cm or less are referred to as microcarcinomas (Berger et al, 1998). These small tumors show a prevalence up to 35% (Harach et al, 1985) at autopsy of individuals who died of causes other than thyroid cancer.
Papillary microcarcinomas are usually found incidentally in thyroidectomy specimens and have a better prognosis than clinically significant PTCs (Lang et al, 1988). In two large series of 190 and 179 patients, only 3 developed metastases and died of the disease and 2 had nodal recurrences, respectively (Sugitani et al, 1998; Tourniaire et al, 1998). However, these tumors have the same histological features as PTCs: enlarged nuclei, thickening of nuclear membrane, nuclear “holes”, and overlapped and grooved nuclei with “ground glass” appearance. It is difficult to assess whether a papillary microcarcinoma is associated with good prognosis or an unfavorable outcome for the patient.
Little is known about the molecular events involved in the development of papillary microcarcinomas. The most frequent molecular alteration in clinically evident PTCs involves the RET proto-oncogene, which maps to the long arm of chromosome 10 at band q11.2. This gene encodes a tyrosine kinase receptor whose expression and function are normally restricted to a subset of cells derived from the neural crest. Activation of the RET oncogene in papillary carcinoma occurs by chromosomal rearrangements that result in the fusion of the RET tyrosine kinase coding region to the aminoterminal coding region of genes that are constitutively expressed in the thyroid (Pasini et al, 1996). Different types of rearrangements have been identified. RET/PTC1 is a paracentric inversion of the long arm of chromosome 10 that juxtaposes the tyrosine kinase (TK) domain of RET to the aminoterminal domain of the gene H4, whose function is unknown (Grieco et al, 1990). RET/PTC2 originates from a translocation involving chromosomes 10 and 17 that results in the fusion of RET and RIα (regulatory subunit of the cAMP-dependent protein kinase A) (Bongarzone et al, 1993). Another paracentric inversion, RET/PTC3, generates a fusion between RET and ELE 1 (Bongarzone et al, 1994). RET/PTC4 is a variant of RET/PTC3 (Fugazzola et al, 1996). Other types of rearrangements have recently been characterized: RET/PTC5, RET/PTC6, RET/PTC7, and RET/KTN1 were all found in children exposed to radioactive fallout after Chernobyl (Klugbauer et al, 1998; Klugbauer and Rabes, 1999; Salassidis et al, 2000), and RET/ELKS (Nakata et al, 1999) and RET/PCM-1 have been detected in our laboratory (Corvi et al, 2000).
The hypothesis that microcarcinoma would represent the early stage of PTC has repeatedly been proposed in the literature (Tallini et al, 1998; Viglietto et al, 1995). However, only a few microcarcinomas seem to develop into PTC, and this is the main reason for the current controversy about the strategy for optimal therapeutic treatment. The aim of this study is to elucidate the role of RET in microcarcinomas and to establish whether RET rearrangements may serve as markers for prognosis. To this end, we used interphase fluorescence in situ hybridization (FISH) to detect rearrangements of RET in paraffin-embedded tissue sections of microcarcinomas. The same technique has been previously applied to study the status of RET in a series of sporadic and familial PTCs (Cinti et al, 2000; Corvi et al, 2001). The FISH approach, based on the analysis at the single-cell level, allows the direct correlation between the histopathological features and the distribution of RET rearrangements in the tumor cells.
Results
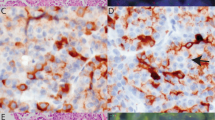
RET rearrangements in microcarcinomas were analyzed by dual color FISH using a probe that encompasses the RET gene along with an α-satellite probe that specifically recognizes centromere 10. In normal thyroid follicular cells, fluorescence microscopy revealed two signals corresponding to the RET gene associated with the two chromosome 10 centromere signals. The tumors that carried RET/PTC1 or RET/PTC3 rearrangements showed one signal associated with one of the centromeres and two signals in the proximity of the homologous centromere (Fig. 1, c and d). In the case of RET translocations (eg, RET/PTC2), the third signal was randomly distributed in the nuclei (data not shown). In cells that carried a trisomy for chromosome 10, the three RET signals were coupled with the three centromere 10 signals (Fig. 1e).

Histopathological and fluorescence in situ hybridization (FISH) analysis of microcarcinomas. a to b, Hematoxylin and eosin staining of a microcarcinoma (×100 and ×400, respectively). c to e, Status of the proto-oncogene RET in thyroid microcarcinoma nuclei. RET cosmids were labeled with biotin and detected with fluorescein-conjugated avidin (green), whereas the α-satellite DNA for centromere 10 was labeled with digoxigenin and detected with anti-digoxigenin-rhodamine Fab fragment (red). c to d, Microcarcinomas showing an inversion involving the RET gene. Filled arrow indicates the rearranged copy of RET (inversion), and the empty arrow indicates nuclei carrying two apparently normal copies of RET. e, Nucleus harboring chromosome 10 trisomy. f, Nuclei of microcarcinoma no. 21 hybridized with α-satellite DNA for centromere 6 (green) and centromere 12 (red) are trisomic for both chromosomes.
Histopathological Analysis
A histopathological analysis of a serial tumor section from each sample was carried out in parallel to FISH to accurately characterize the morphology and size of the microcarcinoma and of the surrounding areas (Fig. 1, a and b). We analyzed 21 papillary microcarcinomas. The age and sex of the patients and morphological features of the samples are reported in Table 1. The area surrounding the tumors was apparently normal in 10 cases only. Lymphocytic thyroiditis was present in three cases, follicular adenoma in three cases, and hyperplasia in the remaining samples. Moreover, this analysis was necessary to closely correlate the hybridization data with the histopathological data from each area of the tissue section.
Assessment of the Cutoff Value
The frequency of nuclei carrying an apparent rearrangement was calculated in each microcarcinoma in the surrounding normal areas of the samples and in two healthy control thyroids (Table 2). To determine the cutoff frequency of rearrangements corresponding to the presence of a real rearrangement, we compared each tumor with the “historical” control obtained by pooling all of the data from the healthy tissues (the two healthy controls and the healthy surrounding areas). In this case the χ2 test was applied with Bonferroni’s correction (p < 0.0024) in view of the multiple comparisons performed. By comparing the data from healthy and tumor samples, we determined a cutoff value of 6.8% (p = 0.0020) corresponding to the presence of a real rearrangement.
FISH Results
RET rearrangements were found in 11 tumors out of 21 (52%) and were absent in all peritumoral tissues and normal thyroid controls. Among the positive tumors, nine carried an inversion and two carried a translocation (Table 2). To determine if these two translocations involved the RET/PTC2 rearrangement, we cohybridized the samples with centromere 17 and RET. A rearrangement-type RET/PTC2 was present in one of the tumors (t.11) and was excluded in the other (t.14), suggesting that this latter tumor may carry a novel type of rearrangement.
Two microcarcinomas (t.12 and t.21), both surrounded by adenomas, carried three copies of RET and three copies of chromosome 10 centromere (Fig. 1e). Subsequent hybridization with α-satellite probes for centromere 6 and 12 confirmed trisomies for these chromosomes as well (Fig. 1f). Parallel histopathological analysis of the two samples showed that trisomies were present both in the microcarcinoma and in the follicular adenoma surrounding it.
Among the three samples that also showed a thyroiditis, only one (t.2) carried a rearrangement. The distribution of rearrangements was heterogeneous among the tumors. Five samples showed a higher percentage of positive cells. Interestingly, these tumors were also characterized by prominent papillae and very marked PTC nuclear features. No significant association was found between the presence of rearrangements and histological and clinical parameters (size, histological variant, lymphocytic infiltration, encapsulation, histology of the tissue surrounding the tumor, presence of metastases, and age of the patient).
Within our series only one patient (no.16) had an aggressive tumor and developed lung metastasis. This tumor did not show any abnormalities involving the RET chromosomal region.
Frequency of Rearrangements in Microcarcinomas versus PTCs
Finally, we compared the frequency of RET rearrangements in microcarcinomas with the frequency found in a series of clinically evident PTCs previously analyzed using the same approach in our laboratory (Cinti et al, 2000). As in the case of the microcarcinomas, we considered positive those PTCs carrying more than 6.8% of cells with the rearrangement. RET rearrangements were significantly more common in microcarcinomas (52% versus 27%, p = 0.03).
Discussion
The common presence of RET rearrangements in microcarcinomas detected in this study confirms the involvement of this oncogene in early stages of thyroid tumorigenesis. Most interestingly, RET rearrangements were significantly prevalent in microcarcinomas in comparison with clinically evident PTCs. These results are in agreement with two other studies that reported 42% and 77% of rearrangements in occult papillary microcarcinomas, respectively (Sugg et al, 1998; Viglietto et al, 1995). Both studies were carried out using RT-PCR with primers for RET/PTC1, RET/PTC2, and RET/PTC3. In the present study, the use of an in situ technique for the detection of RET changes at the DNA level presents several advantages. First, it allows the detection of RET abnormalities, regardless of the type of rearrangement, with the possibility of identifying novel RET fusion genes (Corvi et al, 2000), as in the case of the microcarcinoma carrying the translocation that was different from RET/PTC2. Second, FISH on paraffin-embedded tissue sections allows a strict correlation with different histological parameters and the possibility of analyzing not only tumoral tissue, but also the different areas that surround the tumor within a single experiment. Third, it allowed the detection of changes such as trisomies in some of the samples.
Abundant RET/PTC expression, detectable after RT-PCR by ethidium bromide staining alone without need of hybridization, has been observed in tumors of patients with low-risk clinical parameters and lymphatic involvement (Sugg et al, 1996). Using FISH, which allows a more precise quantification of the relative number of positive cells carrying the rearrangement in the different tumors, we observed that the percentage of detectable cells with rearranged RET was higher in tumors bearing prominent papillae and pronounced nuclear inclusions and groovings. Furthermore, positive cases for RET activation were associated with tumors characterized by prominent papillary areas or small tumor size (Lam et al, 1998; Tallini et al, 1998). Altogether, these data suggest that RET alterations may play an important role in determining the tumor phenotype in vivo.
Although it has been proposed that follicular adenomas represent the early stage of follicular carcinomas, there is no corresponding clear-cut benign state in papillary carcinogenesis. Pathological evidence strongly suggests that the earliest lesion of PTC is the microcarcinoma. Furthermore, from a molecular point of view, RET rearrangements are common features of PTC and microcarcinomas, thus suggesting that the microcarcinoma might be the precursor of the PTC. However, the detection of RET rearrangements in a few adenomas (Cinti et al, 2000; Ishizaka et al, 1991) and the occasional occurrence of PTCs or microcarcinomas within adenomas or adenomatous goiters remain unclear.
Our observation and the evidence that RET/PTC1 oncogene is able to initiate in vivo thyroid tumors support the concept that RET activation is an early event in thyroid carcinogenesis and that further or concomitant molecular events are necessary for neoplastic progression. Soares and collaborators (Soares et al, 1998) described PTC harboring the RET rearrangement as slow-growing papillary tumors that do not usually progress toward less differentiated neoplasms (Tallini et al, 1998). This is also supported by the slowly progressive tumors that develop in the RET/PTC1-transgenic mice (Santoro et al, 1996).
However, an interesting question still to be addressed is why this gene seems to be more frequently activated in microcarcinomas than in clinically evident tumors. From a mechanistic point of view, it is unlikely that the RET rearrangement is deleted and lost during tumor evolution. On the other hand, the major genes responsible for the tumorigenesis of PTCs are probably still unknown. We might envisage that additive and synergistic effects of RET and these genes may play a role in a subgroup of tumors because of the relatively low frequency of RET rearrangements in PTCs. The recent report from Bunone et al (2000) supports the idea that normal thyroid follicular cells can express a functional RET receptor, which may be activated in the presence of specific ligands in the thyroid microenvironment. Therefore, this new concept indicates that the chromosomal rearrangement does not promote the expression of a gene, which is normally completely silent, but leads to the activation of the RET TK, which is no more regulated by external factors. Another interesting notion is that of RET as a dependence receptor (Bordeaux et al, 2000). Receptors like RET and DCC1 (Mehlen et al, 1998) show two distinct forms of signal transduction, depending on their respective ligand availability. In the presence of their ligands, they transduce a signal for either proliferation or differentiation. However, in the absence of their ligands, they induce an active signal for cell death. Therefore, one can speculate that RET rearrangements lead, on one side, to the constitutive activation of RET TK and, on the other, to a modification of the apoptotic activity of RET in thyroid cells. Further efforts must be aimed at clarifying the role of RET activation in follicular cells.
Material and Methods
Samples
Twenty-one cases of papillary microcarcinoma of the thyroid were collected from the pathology files of the St. Bartholomew’s Hospital of London, United Kingdom, (13 cases) and the Arcispedale S. Maria Nuova of Reggio Emilia, Italy (8 cases). Two cases of normal thyroid tissue collected from the latter institution served as controls. All samples were formalin-fixed paraffin-embedded, and two sequential 4-μm-thick sections of each case were routinely stained with hematoxylin and eosin and analyzed by FISH, respectively. The histological analysis also allowed us to measure and precisely localize the microcarcinoma on the slides.
Tissue Sample Preparation for FISH
Tissue sections were dewaxed in xylene and rehydrated in an ethanol series. Pretreatment of the slides was carried out according to the tissue kit (S1337; ONCOR, Inc., Gaithersburg, Maryland) manufacturer protocol. Incubation time for digestion differed for each section, as these differed in their cellularity and thickness.
Probes
A mixture of three cosmids (E5, F2, and R1) covering about 100 kb of the RET genomic region (Pasini et al, 1995) was used. We used a chromosome 10 α-satellite probe as internal control, because the RET proto-oncogene maps on the pericentromeric region of chromosome 10. In addition, we used α-satellite probes specific for chromosomes 17, 12, and 6. The RET probe was labeled with biotin-16-deoxyuridine triphosphate (dUTP), whereas α-satellite probes were labeled with digoxigenin (Roche, Mannheim, Germany) by nick translation (Langer et al, 1981).
Fluorescence In Situ Hybridization
FISH on interphase nuclei was performed according to the protocol previously described (Corvi et al, 1994). The probe concentration was 15 ng/μl for the pool of the RET cosmids. Repetitive sequences for RET were suppressed with 10-fold excess of Cot-1 (Roche). Biotinylated probes were detected with FITC-conjugated avidin (Vector Laboratories, Burlingame, California), and signals were amplified with biotinylated anti-avidin (Vector Laboratories). The digoxigenin-labeled α-satellite probes were detected with rhodamine-conjugated anti-digoxigenin, Fab fragment (Roche). Nuclei were counterstained with 1 μg/ml of 4,6-diamidino-2 phenylindole dihydrochloride and mounted in Vectashield (Vector Laboratories).
Microscopy Analysis
After FISH, the slides were analyzed on a Zeiss Axiophot epifluorescence microscope (Carl Zeiss GmbH, Jena, Germany). The following criteria were followed for the microscopic evaluation of the samples: nuclei had intact morphology, they did not overlap, both RET and centromeric signals were visible, and signals were only counted when they were completely separated from one another. Approximately 100 nuclei were analyzed for each sample.
Statistical Analysis
Statistical analysis was carried out using the χ2 test. To determine the cutoff frequency of rearrangements corresponding to the presence of a real rearrangement, we compared each tumor with the “historical” control, obtained by pooling all of the data from the normal tissues. In this case, the χ2 test was applied with Bonferroni’s correction (p < 0.0024) in view of the multiple comparisons performed (Bland, 2000).
References
Berger N, Borda A, and Bizollon MH (1998). Thyroid papillary carcinoma and its variants. Arch Anat Cytol Pathol 46: 45–61.
Bland M (2000). Multiple significance tests and the Bonferroni correction. In: An introduction to medical statistics. New York: Oxford University Press, 121–142.
Bongarzone I, Butti MG, Coronelli S, Borrello MG, Santoro M, Mondellini P, Pilotti S, Fusco A, Della Porta G, and Pierotti MA (1994). Frequent activation of ret proto-oncogene by fusion with a new activating gene in papillary thyroid carcinomas. Cancer Res 54: 2979–2985.
Bongarzone I, Monzini N, Borrello MG, Carcano C, Ferraresi G, Arighi E, Mondellini P, Della Porta G, and Pierotti MA (1993). Molecular characterization of a thyroid tumor-specific transforming sequence formed by the fusion of ret tyrosine kinase and the regulatory subunit RI alpha of cyclic AMP-dependent protein kinase A. Mol Cell Biol 13: 358–366.
Bordeaux MC, Forcet C, Granger L, Corset V, Bidaud C, Billaud M, Bredesen DE, Edery P, and Mehlen P (2000). The RET proto-oncogene induces apoptosis: A novel mechanism for Hirschsprung disease. EMBO J 19: 4056–4063.
Bunone G, Uggeri M, Mondellini P, Pierotti MA, and Bongarzone I (2000). RET receptor expression in thyroid follicular epithelial cell-derived tumors. Cancer Res 60: 2845–2849.
Cinti R, Yin L, Ilc K, Berger N, Basolo F, Cuccato S, Giannini R, Torre G, Miccoli P, Amati P, Romeo G, and Corvi R (2000). RET rearrangements in papillary thyroid carcinomas and adenomas detected by interphase FISH. Cytogenet Cell Genet 88: 56–61.
Corvi R, Amler LC, Savelyeva L, Gehring M, and Schwab M (1994). MYCN is retained in single copy at chromosome 2 band p23–24 during amplification in human neuroblastoma cells. Proc Natl Acad Sci USA 91: 5523–5527.
Corvi R, Berger N, Balczon R, and Romeo G (2000). RET/PCM-1: A novel fusion gene in papillary thyroid carcinoma. Oncogene 19: 4236–4242.
Corvi R, Lesueur F, Martinez-Alfaro M, Zini M, Decaussin M, Murat A, and Romeo G (In press, 2001). RET rearrangements in familial papillary thyroid carcinomas. Cancer Lett 170:191–198.
Fugazzola L, Pierotti MA, Vigano E, Pacini F, Vorontsova TV, and Bongarzone I (1996). Molecular and biochemical analysis of RET/PTC4, a novel oncogenic rearrangement between RET and ELE1 genes, in a post-Chernobyl papillary thyroid cancer. Oncogene 13: 1093–1097.
Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G, Fusco A, and Vecchio G (1990). PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell 60: 557–563.
Harach HR, Franssila KO, and Wasenius VM (1985). Occult papillary carcinoma of the thyroid. A “normal” finding in Finland. A systematic autopsy study. Cancer 56: 531–538.
Ishizaka Y, Kobayashi S, Ushijima T, Hirohashi S, Sugimura T, and Nagao M (1991). Detection of retTPC/PTC transcripts in thyroid adenomas and adenomatous goiter by an RT-PCR method. Oncogene 6: 1667–1672.
Klugbauer S, Demidchik EP, Lengfelder E, and Rabes HM (1998). Detection of a novel type of RET rearrangement (PTC5) in thyroid carcinomas after Chernobyl and analysis of the involved RET-fused gene RFG5. Cancer Res 58: 198–203.
Klugbauer S and Rabes HM (1999). The transcription coactivator HTIF1 and a related protein are fused to the RET receptor tyrosine kinase in childhood papillary thyroid carcinomas. Oncogene 18: 4388–4393.
Lam AK, Montone KT, Nolan KA, and Livolsi VA (1998). Ret oncogene activation in papillary thyroid carcinoma: Prevalence and implication on the histological parameters. Hum Pathol 29: 565–568.
Lang W, Borrusch H, and Bauer L (1988). Occult carcinomas of the thyroid. Evaluation of 1,020 sequential autopsies. Am J Clin Pathol 90: 72–76.
Langer PR, Waldrop AA, and Ward DC (1981). Enzymatic synthesis of biotin-labeled polynucleotides: Novel nucleic acid affinity probes. Proc Natl Acad Sci USA 78: 6633–6637.
Mehlen P, Rabizadeh S, Snipas SJ, Assa-Munt N, Salvesen GS, and Bredesen DE (1998). The DCC gene product induces apoptosis by a mechanism requiring receptor proteolysis. Nature 395: 801–804.
Nakata T, Kitamura Y, Shimizu K, Tanaka S, Fujimori M, Yokoyama S, Ito K, and Emi M (1999). Fusion of a novel gene, ELKS, to RET due to translocation t(10;12)(q11;p13) in a papillary thyroid carcinoma. Genes Chromosom Cancer 25: 97–103.
Parkin DM, Pisani P, and Ferlay J (1999). Estimates of the worldwide incidence of 25 major cancers in 1990. Int J Cancer 80: 827–841.
Pasini B, Ceccherini I, and Romeo G (1996). RET mutations in human disease. Trends Genet 12: 138–144.
Pasini B, Hofstra RM, Yin L, Bocciardi R, Santamaria G, Grootscholten PM, Ceccherini I, Patrone G, Priolo M, and Buys CH (1995). The physical map of the human RET proto-oncogene. Oncogene 11: 1737–1743.
Salassidis K, Bruch J, Zitzelsberger H, Lengfelder E, Kellerer AM, and Bauchinger M (2000). Translocation t(10;14)(q11.2: q22.1) fusing the kinetin to the RET gene creates a novel rearranged form (PTC8) of the RET proto-oncogene in radiation-induced childhood papillary thyroid carcinoma. Cancer Res 60: 2786–2789.
Santoro M, Chiappetta G, Cerrato A, Salvatore D, Zhang L, Manzo G, Picone A, Portella G, Santelli G, Vecchio G, and Fusco A (1996). Development of thyroid papillary carcinomas secondary to tissue-specific expression of the RET/PTC1 oncogene in transgenic mice. Oncogene 12: 1821–1826.
Soares P, Fonseca E, Wynford-Thomas D, and Sobrinho-Simoes M (1998). Sporadic ret-rearranged papillary carcinoma of the thyroid: A subset of slow growing, less aggressive thyroid neoplasms? J Pathol 185: 71–78.
Sugg SL, Ezzat S, Rosen IB, Freeman JL, and Asa SL (1998). Distinct multiple RET/PTC gene rearrangements in multifocal papillary thyroid neoplasia. J Clin Endocrinol Metab 83: 4116–4122.
Sugg SL, Zheng L, Rosen IB, Freeman JL, Ezzat S, and Asa SL (1996). Ret/PTC-1, -2, and -3 oncogene rearrangements in human thyroid carcinomas: Implications for metastatic potential? J Clin Endocrinol Metab 81: 3360–3365.
Sugitani I, Yanagisawa A, Shimizu A, Kato M, and Fujimoto Y (1998). Clinicopathologic and immunohistochemical studies of papillary thyroid microcarcinoma presenting with cervical lymphadenopathy. World J Surg 22: 731–737.
Tallini G, Santoro M, Helie M, Carlomagno F, Salvatore G, Chiappetta G, Carcangiu ML, and Fusco A (1998). RET/PTC oncogene activation defines a subset of papillary thyroid carcinomas lacking evidence of progression to poorly differentiated or undifferentiated tumor phenotypes. Clin Cancer Res 4: 287–294.
Tourniaire J, Bernard MH, Bizollon-Roblin MH, Bertholon-Gregoire M, and Berger-Dutrieux N (1998). Papillary microcarcinoma of the thyroid. 179 cases reported since 1973. Presse Med 27: 1467–1469.
Viglietto G, Chiappetta G, Martinez-Tello FJ, Fukunaga FH, Tallini G, Rigopoulou D, Visconti R, Mastro A, Santoro M, and Fusco A (1995). RET/PTC oncogene activation is an early event in thyroid carcinogenesis. Oncogene 11: 1207–1210.
Acknowledgements
This work was supported by the Comité Départemental du Rhône de la Ligue Nationale contre le Cancer, the Association pour la Recherche sur le Cancer, and the International Agency for Research on Cancer.
We are grateful to Mrs. S. Pauly for technical assistance and to Dr. P. Taniere for critical discussions and review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
RC and MM-A contributed equally to this work.
Rights and permissions
About this article
Cite this article
Corvi, R., Martinez-Alfaro, M., Harach, H. et al. Frequent RET Rearrangements in Thyroid Papillary Microcarcinoma Detected by Interphase Fluorescence In Situ Hybridization. Lab Invest 81, 1639–1645 (2001). https://doi.org/10.1038/labinvest.3780377
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780377
This article is cited by
-
Pathogenetic mechanisms in thyroid follicular-cell neoplasia
Nature Reviews Cancer (2006)
-
Ras-mediated apoptosis of PC CL 3 rat thyroid cells induced by RET/PTC oncogenes
Oncogene (2003)
