
Abstract
Placental site trophoblastic tumor (PSTT) is a neoplastic proliferation of intermediate trophoblasts that invades the myometrium at the placental site after a pregnancy. Less than 100 cases have been reported. Information of the sex assignment of the antecedent gestation is available in 21 cases: 18 of these were female. To explore this interesting phenomenon, we have determined the sex chromosome composition of the tumor tissue preserved in paraffin blocks for five new cases of this condition. The last documented gestational event included a normal vaginal delivery of female infants in three cases, normal vaginal delivery of an infant of unknown sex in one case and a molar gestation in one case. Using the X-linked human androgen receptor (AR) gene as a polymorphic marker, we showed that in all five cases the tumor had a likely XX chromosomal composition; and in four cases it was possible to determine that one of the X chromosomes was of paternal origin. In one case, the paternal X chromosome showed no polymorphism to either maternal X chromosomes. In addition, sensitive semi-nested PCR failed to show a human Y chromosome element in any of the five cases of PSTT. Overall, of 21 cases from the literature and 5 cases of ours, 89% (23 of 26) showed an XX genomic composition in PSTT, either by history or genetic analysis. These results suggest that most PSTT were derived from the antecedent female conceptus and were likely to have possessed a functional paternal X chromosome. Methylation status analysis at the AR locus was performed in the three PSTT in which the paternal X chromosome was identifiable. In two cases, the paternal AR locus was hypomethylated while the corresponding maternal locus was hypermethylated. The methylation status of other loci was not investigated. Collectively, sex chromosome analysis of five cases of PSTT with literature support suggests a unique genetic basis for the development of PSTT that involves the paternal X chromosome. Although largely speculative, an active paternal X chromosome may be of importance in the pathogenesis of PSTT.
Similar content being viewed by others

Introduction
During conception, tissue having paternally-derived genetic material is implanted in the female uterus. Rarely, this event leads to the development of a disease in the mother in the form of gestational trophoblastic disease (GTD). GTD includes some entities characterized by distinct unique genetic modes of pathogenesis which have been recognized for long time (Kajii and Ohama, 1977; Szulman, 1984): in complete hydatidiform moles, more than 90% of the proliferative trophoblasts have a 46, XX diploid pattern, all derived from the sperm (androgenesis). In partial moles, the karyotype is triploid in most cases (eg, 69, XXY), resulting from fertilization of an egg with one or two sperm. In fact, triploid gestations in which there is no duplication of the paternal chromosomes do not have trophoblastic hyperplasia and do not meet the histologic criteria of molar gestation (Jacobs et al, 1982). Therefore, duplication of the paternal X chromosome with or without the maternal X chromosome seems to result in abnormal genetic overdosing from the paternal X chromosome and seems to play a role in trophoblastic proliferation. A distinct genetic mode has not yet been identified in other trophoblastic lesions such as placental site trophoblastic tumor (PSTT) and epitheloid trophoblastic tumor. Recently, evidence indicates there are X chromosome-linked, genetically imprinted genes that specifically regulate the development of extraembryonic tissues (Li et al, 1997; Lin et al, 1994) and that the X chromosome inactivation in an XX embryo is crucial to the development of normal extraembryonic tissue (McGrath and Solter, 1984). In mouse, the paternally derived X chromosome is preferentially inactivated in the primitive trophectoderm cell lineages (Takagi and Sasaki, 1975). Evidence exists to suggest that this may be true in human as well (Goto et al, 1997), although it is debatable. Although the purpose and mechanism of genomic imprinting, including X chromosome inactivation, is not well understood, epigenetic regulation of gene expression through imprinting and its implication in tumorigenesis has drawn great attention recently. Somatic chromosomal imprinting studies suggest that uniparental gene expression can function as a cancer predisposing and/or initiation event (Pulford et al 1999).
PSTT is an infiltrating tumor of the intermediate trophoblasts (Young et al, 1988). This tumor predominantly consists of mononucleated cells with voluminous cytoplasm. The tumor cells are positive for human placental lactogen but rarely for human chorionic gonadotropin. Most cases of PSTT pursue a benign clinical course; however, malignant cases with an estimated mortality rate of 15% have been reported. Treatment of PSTT is largely surgical because it is not usually sensitive to chemotherapy. The risk factors and genetic requirement for PSTT, and its pathogenesis are largely unknown. In this report, we have explored the histogenesis of PSTT by analyzing the sex chromosome composition and status of X chromosome inactivation in five new cases of PSTT. With additional support from the literature, we suggest that the development of PSTT enroutes a unique genetic mode requiring the presence of a paternal X chromosome.
Results
Literature Review
Less than 100 cases of PSTT were identified in the literature, and only 72 cases had detailed clinical and pathology information. A recent thorough literature review on PSTT can be found elsewhere (Chang et al, 1999) and therefore, only the information regarding the gender of antecedent gestation is reviewed here (Table 1).
The sex of the antecedent gestation is known in 21 cases. Of these, 18 had a female antecedent gestation by history, this was confirmed by DNA analysis in 4 cases (Fisher et al, 1992; Kodama et al, 1996; Lathrop et al, 1988). In one case, in which the preceding gestational event was a complete mole, the DNA analyses confirmed that the tumors harbored two X chromosomes (Fisher et al, 1992). Only two patients developed PSTT with antecedent delivery of male infants (Dessau et al, 1990; Hoffman et al, 1993). A single case was reported that suggested the presence of a Y chromosome upon genetic analysis, although the antecedent gestation was female and Southern blots using five autosomal RFLP markers showed that the tumor DNA had identical hybridization patterns to DNA from the patient’s second daughter (Arima et al, 1994, 1995).
Clinical and Pathological Information of Five New Cases of PSTT
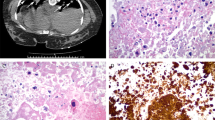
The clinical information on these cases is included in Table 1. The age of the patients at presentation ranged between 30 and 48 years. Gravidity ranged between 2 and 4. Three women presented with abnormal vaginal bleeding 1 to 6 years after their last documented gestational event, which was a normal vaginal delivery of a female infant in three cases, a normal vaginal delivery of a infant of unknown sex in one case, and a molar gestation in one case. Human chorionic gonadotrophin values ranged between 190 to 1600 at presentation. All patients underwent abdominal hysterectomy after the diagnosis of PSTT was made after dilatation and curettage. The resected uteri revealed polypoid intrauterine tumor masses (1.8 to 3 cm) grossly invading the myometrium. The histology of all five tumors was similar and was characterized by sheets of intermediate trophoblasts invading the myometrium. In case #5, the preceding complete mole showed typical hydropic swelling with absence of vascularization of villi. Striking proliferation of both cytotrophoblasts and syncytiotrophoblasts was present (Fig. 1).

Representative histology of case #5. T: placental site trophoblastic tumor (PSTT) (H&E × 40), M: normal myometrium adjacent to the PSTT (H&E, × 40), CM: complete mole (H&E, × 20), GE: gestational endometrium associated with complete mole (H&E, × 20).
Sex Chromosome Analysis
We investigated the X chromosome composition using a PCR-based assay of the androgen receptor (AR) locus on the X chromosome (Mutter et al, 1995). We used primers flanking the highly polymorphic trinucleotide repeat (CAG) in the first exon of the human AR gene to detect and distinguish the paternal and maternal X chromosomes (Xm and Xp, respectively). This assay yields an amplified product of 229 to 289 bps, with the size varying from allele to allele, depending on the number of trinucleotide repeats (11 to 31). Using this assay, one can distinguish between different X chromosomes. Because contaminating normal myometrial cells are usually present in PSTT tumors, one could expect to find up to three alleles: two from the PSTT cells and a third allele from contaminating maternal cells, which is distinct from the maternal allele present in the tumor.
Analysis of all five cases revealed the presence of a likely paternal X chromosome (Xp). PCR amplification in cases 1 and 2 showed similar results (Fig. 2, lanes 1 to 4). The normal myometrium showed a single band pattern, in contrast to the double band pattern seen in the tumor. The single band from the myometrium indicated that the two X chromosomes of the mother had no polymorphism between the AR alleles. The tumor cells gave one band identical to the one amplified from the normal myometrium and an additional low molecular weight band on the gel. This indicated that the second band was derived from the paternal X chromosome, supporting the idea that the two PSTT were conceptus-derived and XmXp in chromosomal composition. In case #3, the pattern of PCR products obtained suggested an XX genetic composition in which the origin of the X chromosomes could not be ascertained, as a sample of paternal DNA was not available (Fig. 2, lanes 5 and 6). One possibility is that this tumor was exclusively of maternal origin, but this seems unlikely (see discussion). In case #4, the two maternal AR genes in the normal myometrium were polymorphic, and yielded two bands in PCR analysis. We obtained three AR-derived PCR products from the tumor tissue (Fig. 2, lanes 7 and 8); two bands apparently identical to those in the normal control tissue and an additional higher weight molecular band, undoubtedly of paternal origin. One of the two lower bands could have been derived from contaminating normal maternal tissue. These results suggested that this tumor was XmXp in composition.

X chromosome analyses of PSTT (Case #1 to Case #4): T: PSTT, N: normal myometrium. Lanes 1, 2: Case #1. Lanes 3, 4: Case #2. Lanes 5, 6: Case #3. Lanes 7, 8: Case #4.
Case #5 is of special interest because the PSTT developed after a complete hydatidiform mole. Both tumor and molar tissues were available for analysis (Fig. 1). PCR analysis of the PSTT tumor in case #5 showed three distinct bands (Fig. 3, lane 1). Two were identical to two X chromosomal bands found in the normal myometrium. An intermediate band was present in the PSTT, which was identical to the single band amplified from the previously diagnosed complete mole (Fig. 3, lane 3). Normal corresponding myometrium to PSTT and gestational endometrium to mole yielded two maternal bands (Fig. 3, lanes 2 and 4). There are two possible interpretations for this result. The most likely one is that the mole harbored two identical X chromosomes, both of paternal origin (XpXp), which gave rise to a single band. A similar band was seen in the PSTT, and the remaining two bands were contaminants from normal maternal tissue/myometrium. Therefore, the PSTT likely harbored two X chromosomes, both of paternal origin. Another possibility is that the PSTT arose from a different non-molar female gestation. In this case, one band each would have come from maternal and paternal sources and the remaining band would have been derived from contaminant maternal tissues (similar to case #4). Given the temporal relationship of the molar pregnancy and the PSTT, it is likely that the paternal X chromosome originated from this complete mole, rather than a different gestation, although there were two female term pregnancies before the mole. It is unlikely that a single unique band represented an XY gestation with two contaminant bands, as semi-nested PCR to amplify the Y chromosome-specific target was negative in all five cases (Fig. 4), in contrast to the robust positive amplification (a 307 bp specific product) from two male control DNA samples (Fig. 4, lanes 7 and 8).

X chromosome composition in case #5. T: PSTT (lane 1); N: normal myometrium (lane 2); CM: complete molar tissue (lane 3) and GE: gestational endometrium adjacent to the molar tissue (lane 4).

Detection of Y chromosomal element by semi-nested PCR of PSTT DNA. A strong positive product of 307 bp is present in both positive control specimens, but absent in all PSTT. Lane 1: 100 bp DNA marker. Lanes 2 to 6: PSTT case #1 to 5. Lanes 7 and 8: positive controls using DNA template from two male tissues. Several faint smaller bands in lane 6 are non-specific products.
Methylation Status at AR Alleles
From the data presented above, it seems that a paternal X chromosome is required for the development of PSTT. This implies a functional role in tumorigenesis for gene(s) on the paternal X chromosome. If Xp is required for tumorigenesis, one would predict preferential nonmethylation of the X chromosome, viz., an active Xp. We assessed the methylation status of AR alleles in three of five of our PSTT (in two cases, it was not possible to unambiguously identify the methylation status of PCR product derived from Xp). We predigested the DNA with the methylation-sensitive restriction enzyme, HhaI, and then amplified the DNA using the same primers described above for the 5′ end of the AR gene. Among the three cases analyzed, two cases (case #1 and case #4) showed preferential digestion of the paternal AR allele by HhaI (Fig. 5, A and B, respectively) indicating hypomethylation of Xp. We also saw resistance of the maternal AR allele to HhaI digestion, consistent with inactivation of the maternal X chromosome. In one case (case #2), however, the AR locus seemed to be active on the Xm chromosome but hypermethylated on the paternal Xp chromosome (Fig. 5C).

Methylation status at the AR locus on the X chromosome of PSTT by HUMARA analysis. (+) and (−) signs refer to +/- HhaI digestion. (A) Case #1: Lanes 1 and 2 are PCR products of tumor (T) and lanes 3 and 4 are products of control tissue (N). The paternal band (lower) is present only in tumor cells and is preferentially skewed upon HhaI digestion. (B) Case #4: The tumor cells (T) contribute an additional high molecular paternal band (upper band, lane 1) which does not exist in maternal normal control tissue (N, lanes 3 and 4). This paternal band seems preferentially skewed upon HhaI digestion (lane 2). (C) Case #2: The tumor harbors a unique paternal band (lane 1, lower band). The upper maternal band is preferentially hypomethylated while the lower paternal band is hypermethylated (lane 2, with HhaI digestion). The two normal maternal X chromosomes show no polymorphism at the AR locus (lane 3). The weak signal in normal tissue (lane 3 and 4) reflects severe degradation of the DNA in this case.
Discussion
Tumors included in the category of GTD are unique among neoplasms in their histogenesis and chromosomal complement, and thus represent lesions of special interest. One would expect, a priori, gestational trophoblastic tumors to occur with equal frequency after male and female conceptuses. We present, through combined literature review and five new cases, 26 PSTT, which remarkably seem nearly exclusively to either have an XmXp composition, or to have followed a female conceptus, in which case one would similarly expect an XmXp composition. All five cases of PSTT analyzed here showed an XX chromosomal composition, indicating the origin from an antecedent female conceptus or from an antecedent complete mole. Four other cases of PSTT in the literature had, by molecular analysis, an XmXp chromosomal composition (Fisher et al, 1992; Kodama et al, 1996; Lathrop et al, 1988). Overall, 89% of the 26 cases (23 of 26) showed a XX genomic makeup in PSTT either by history or genetic analysis (Table 1).
Case #3 of our series showed two PCR products in the tumor which were identical in size to the two normal maternal products. We favor the explanation that the paternally derived AR allele has no polymorphism more then that of two maternal AR alleles. It is very unlikely that this tumor was of maternal origin as the tumor was composed of purely intermediate trophoblast-type cells without significant amounts of normal maternal tissue. In addition, a negative result for PCR amplification of Y chromosome DNA (Fig. 4) also argues against the possibility of a XY chromosome composition of a male-conceptus derived tumor.
Case #5, also reported here, is of special interest, in that it occurred after a molar pregnancy, which, in the case of complete moles, has an XpXp composition. Tissue from both the mole and the PSTT were available for analysis (Fig. 1). We identified a paternal AR allele in the molar tissue that was distinct from the two maternal AR alleles (Fig. 3, lanes 3 and 4). Our data suggests that this paternal AR allele was also present in the PSTT cells along with the two maternal AR alleles. This indicates that the PSTT contained at least one AR allele from paternal origin. Whether it developed directly from the antecedent molar tissue or possibly from an earlier female pregnancy cannot be conclusively determined by this study. Given the temporal proximity, it is most likely that the PSTT developed from the most recent molar pregnancy. PSTT have been reported to develop after molar pregnancies (Fisher et al, 1992).
There are important exceptions to the apparent rule of an XX composition in PSTT. One case reported by Arima et al had a possible Y chromosome component in the tumor DNA, analyzed by PCR. However, the patient had two preceding female conceptions and Southern blotting using five autosomal RFLP markers showed that the tumor DNA had identical hybridization patterns to the DNA from the patient’s second daughter (Arima et al, 1994, 1995). These data suggest either a false-positive assay for Y genetic material, or an incomplete reproductive history. One case reported by Dessau et al involved a patient that developed a PSTT 31 months after a term delivery of a male infant (Dessau et al, 1990). Hoffman et al reported a similar situation (Hoffman et al, 1993). However, no genetic or chromosome analyses were available in these cases. Overall, the number of PSTT after male gestations remains surprisingly small.
Considering all of the information available to us, the suggestion could be made that PSTT are derived from the extraembryonic tissue of an antecedent female conceptus. This observation indicates a pathogenesis and genetic origin for PSTT that is unique and likely requires a Xp. The Xp may be involved in this tumor in two possible ways. One is that in these cases Xp harbors a dominant oncogene, although another is that the tumorigenesis results from abnormal dosage of functional X chromosomes. Although no definitive candidate oncogenes exist, possibilities include Esx1, Pem, MYCL2, and IAP (Deveraux et al, 1997; Li et al, 1997; Lin et al, 1994; Redolfi et al, 1999). Further studies are needed to substantiate the involvement of any of these genes in PSTT. However, because PSTT is an extremely rare neoplasm while female conceptuses harboring an Xp are extremely common, the hypothesis of a dominant oncogene on Xp would require either mutational activation of such a gene, or one or more cooperating oncogenic events. In addition, there exists no published data to support a familial susceptibility to PSTT. The contention that the Xp of PSTT harbors a dominant oncogene would require that this X chromosome be active.
Distinct unique genetic modes of pathogenesis have been recognized for many years in hydatidiform moles, eg, the presence of an Xp has been shown in most molar chromosome compositions. PSTT is perhaps another GTD whose pathogenesis requires an Xp. Unlike hydatidiform mole, PSTT is a true neoplasm, which carries a definite malignant potential (Fukunaga and Ushigome, 1993). The unique chromosome composition of PSTT may reflect a role for abnormal gene dosage in tumorigenesis. It has been demonstrated that the paternal genome is essential for the invasive and proliferative capacity of the placenta during early implantation (Barlow, 1995). During the past decade, tumorigenic genes have been discovered to transit from functional haploidy to diploidy through loss of imprinting (Glassman et al, 1996). Among autosomal genes, examples include H19 and IGF2 (Jinno et al, 1995), which are paternally imprinted in extraembryonic tissue. This imprinting seems to be relaxed in trophoblast-derived choriocarcinoma and hydatidiform mole (Ariel et al, 1994; Rachmilewitz et al, 1995; Walsh et al, 1995). Relaxation of X chromosome imprinting, however, has not heretofore been implicated in tumorigenesis. Several groups have shown that, as in mouse, human female trophectoderm shows preferential inactivation of the paternal X (Goto et al, 1997; Harrison, 1989; Harrison and Warburton, 1986; Ropers et al, 1978). Others have shown the opposite observation, where the inactivation of X chromosome is heterogeneous and random, which is supported by the most recent report (Looijenga et al, 1999). Before this issue is finally settled, the presence (two of three cases) of an active (hypomethylated) AR locus of paternal X chromosome in our limited study is of great interest and requires further study of more PSTT cases. There is evidence that the relaxation of genomic imprinting can be regional along a chromosome. This may explain the methylation data of our third case where the paternal AR locus was methylated and the potentially active region could not be reached by our methodology. Although largely speculative, either the relaxation of Xp chromosome inactivation or simply double dosing of the X chromosome in trophoblastic cells may lead to abnormal proliferation, such as seen in hydatidiform mole and PSTT. Methylation studies of X chromosome inactivation of more cases with multiple markers are important to reach a conclusion.
Materials and Methods
Literature Review
MEDLINE search for PSTT was performed. Less than 100 cases were found in the literature. Among these, approximately 70 cases were found with detailed clinical data, but the sex assignment of antecedent gestation was available in only 21 cases.
Case Review
All five cases of PSTT were retrieved from the surgical pathology files at Yale-New Haven Hospital, New Haven, CT. Dilation & curettage and hysterectomy specimens were available for review in all cases. H&E-stained sections of formalin-fixed paraffin-embedded tissue were reviewed in all cases and selected slides were stained for human chorionic gonadotropin (Dilution 1:3200; Dako, Carpinteria, California) and human placental lactogen (Dilution 1:12,800; Dako).
Tissue Selection and DNA Extraction
Formalin-fixed paraffin-embedded tissue blocks, from both the tumor and the adjacent normal myometrium were obtained from each case. Tissue sections with well-demarked tumor areas were selected, and 5 to 10 μm-thick paraffin sections were prepared. Approximately 10 to 20 μg (1000 to 2000 cells) of tissue was obtained from each sample. DNA was then extracted by using Qiagen Tissue DNA preparation kit (QiamKit; Qiagen, Studio City, California) according to the manufacturer’s instructions. The DNA was eluted into 30 μl of water.
Sex Chromosome and Methylation Status Analyses
PCR amplification of human AR gene was carried out by amplification of DNA with primers Hum-1 and Hum-2 (5′-CCG AGG AGC TTT CCA GAA TC-3′ and 5′-TAC GAT GGG CTT GGG GAG AA-3′, respectively) in a 50 μl PCR reaction (10 mmol/L Tris, pH8.4; 50 mmol/L KCL; 20 mg/ml of gelatin; 1.5 mmol/L MgCl2; primers 0.3 mmol/L each; 0.2 mmol/L dATP; 0.2 mmol/L 7-deaza-dGTP; 0.2 mmol/L dTTP; 0.05 mmol/L dCTP); 0.2 μCi/50 ul reaction of α-32P-dCTP was added to the reaction mix to label amplified products. The PCR began with a “ hot start” at 97° C for 8 minutes, followed by a 4 minutes dwell at 85° C, during which 1.25 units Taq polymerase (Boehringer Mannheim, Mannheim, Germany) was added. The reaction mixture went through 3 cycles of prolonged denaturation at 95° C (4 minutes), annealing at 55° C (120 seconds) and primer extension at 72° C (90 seconds). The reaction then received 30 cycles of 95° C (30 seconds), 55° C (45 seconds) and 72° C (90 seconds). The final PCR product was incubated at 72° C for 15 minutes. The products were subjected to electrophoresis at 300 V overnight in an 8% non-denaturing 0.5 mm-thick polyacrylamide gel made in 1x TBE buffer. Gel was dried and autoradiographed with Kodak XAR film at -70° C. DNA methylation status of the AR allele was performed with pre-digestion of DNA with HhaI enzyme for 3 hours at 37° C before PCR analysis.
To confirm the presence or absence of an Y chromosomal contribution to PSTT, semi-nested PCR using Y chromosome-specific (ZFY gene) oligos was performed to amplify a 307 bp product (Liou et al, 1993). Three microliters of DNA extract was amplified using first-pair primers (5′-CCA TTT GTT CTA AGT CGC CA-3′ and 5′-GCT TGT AGA CAC ACT GTT AGG-3′) in 50 μl of PCR reaction for 30 cycles at 95° C (1 minute), annealing at 55° C (1 minute) and primer extension at 72° C (1 minute). One microliter of PCR reaction was subject to a second nested PCR amplification using 5′-CTC TCA GTT CAC ACA AAG G-3′ and 5′-GCT TGT AGA CAC ACT GTT AGG-3′) with 40 cycles at similar PCR conditions. Fifteen μl of PCR products were loaded onto 1.5% agarose gels. Positive controls used male tissue (two cases of synovial sarcoma) as source of DNA template.
References
Ariel I, Lustig O, Oyer CE, Elkin M, Gonik B, Rachmilewitz J, Biran H, Goshen R, DeGroot N, and Hochberg A (1994). Relaxation of imprinting in trophoblastic disease. Gynecol Oncol 53:212–219.
Arima T, Imamura T, Amada S, Tsuneyoshi M, and Wake N (1994). Genetic origin of malignant trophoblastic neoplasms. Cancer Genet Cytogenet 73:95–102.
Arima TI, Sakuragi N, Higashi M, Kamua T, Fujimoto S, Nakano H, and Wake N (1995). Malignant trophoblastic neoplasms with different modes of origin. Cancer Genet Cytogenet 85:5–15.
Barlow D (1995). Gametic imprinting in mammals. Science 270:1610–1613.
Berkowitz BJ, Jones JG, Merkatz IR, and Runowica CD (1990). Ovarian conservation in placental site trophoblastic tumor. Gynecol Oncol 37:239–243.
Chang Y, Chang TC, Hsueh S, Huang KG, Wang PN, Liu HP, and Soong YK (1999). Prognostic factors and treatment for placental site trophoblastic tumor - report of 3 cases and analysis of 88 cases. Gynecol Oncol 73:216–222.
Dessau R, Rustin GJS, Dend J, Paradinas FJ, and Bagshawe KD (1990). Surgery and chemotherapy in the management of placental site tumor. Gynecol Oncol 39:56–59.
Deveraux QL, Takahashi R, Salvesen GS, and Reed JC (1997). X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388:300–303.
Fisher RA, Paradinas FJ, Newlands ES, and Boxer GM (1992). Genetic evidence that placental site trophoblastic tumours can originate from a hydatidiform mole or a normal conceptus. Br J Cancer 65:355–358.
Fukunaga M and Ushigome S (1993). Malignant trophoblastic tumors: Immunohistochemical and flow cytometric comparison of choriocarcinoma and placental site trophoblastic tumors. Hum Pathol 24:1098–1106.
Glassman M, De Groot N, and Hochberg A (1996). Relaxation of imprinting in carcinogenesis. Cancer Genet Cytogenet 89:69–73.
Gloor E, and Harlimann J (1981). Trophoblastic pseudotumor of the uterus, clinicopathologic report with immunohistochemical and ultrastructural studies. Am J Surg Pathol 5:5–13.
Goto T, Wright E, and Monk M (1997). Paternal X-chromosome inactivation in human trophoblastic cells. Mol Hum Reprod 3:77–80.
Harrison K (1989). X-chromosome inactivation in the human cytotrophoblast. Cytogenet Cell Genet 52:37–41.
Harrison K and Warburton D (1986). Preferential X-chromosome activity in human female placental tissues. Cytogenet Cell Genet 41:163–168.
Hoffman J, Silverman AD, Gelber J, and Catun R (1993). Placental site trophoblastic tumor: A report of radiologic, surgical and pathologic methods of evaluating the extent of disease. Gynecol Oncol 50:110–114.
Jacobs P, Szulman AE, Funkhouser J, Matsuura JA, and Wilson CC (1982). Human triploidy: Relationship between parental origin of the additional haploid complement and development of partial hydatidiform mole. Ann Hum Genet 46:223–231.
Janni W, Hantschmann P, Rehbock J, Braun S, Lochmueller E, and Kindermann G (1999). Successful treatment of malignant placental site trophoblastic tumor with combined cytostatic-surgical approach: Case report and review of literature. Gynecol Oncol 75:164–169.
Jinno Y, Ikeda Y, Yun K, Maw M, Masuzaki H, Fukuda G, Inuzuka K, Fujishita A, Ohtani Y, Okimoto T, Ishimura T, and Niikawa N (1995). Establishment of functional imprinting of the H19 gene in human developing placentae. Nat Genet 10:318–324.
Kajii T and Ohama K (1977). Androgenetic origin of hydatidiform mole. Nature 268:633–634.
Kodama S, Kase H, Aoki Y, Yahata T, Tanaka K, Motoyama T, and Kewa K (1996). Recurrent placental site trophoblastic tumor of the uterus: Clinical, pathologic, ultrastructural, and DNA fingerprint study. Gynecol Oncol 60:89–93.
Lathrop J, Wachtel FTJ, and Meissner GF (1978). Uterine choriocarcinoma fourteen years following bilateral tubal ligaiton. Obstet Gynecol 51:477–482.
Lathrop JL, Lauchlan S, Nayak R, and Ambler M (1988). Clinical characteristics of placental site trophoblastic tumor (PSTT). Gynecol Oncol 31:32–42.
Li Y, Lemaire P, and Behringer RR (1997). Esx1, a novel X chromosome-linked homeobox gene expressed in mouse extraembryonic tissues and male germ cells. Dev Biol 88:85–95.
Liou J, Pao CC, Hor JJ, and Kao SM (1993). Fetal cells in the maternal circulation during first trimester in pregnancies. Hum Genet 92:309–311.
Lin T, Labosky PA, Grabel LB, Kozak CA, Pitman JP, Kleeman J, and MacLeod CL (1994). The Pem homeobox gene is X-linked and exclusively expressed in extraembryonic tissues during early murine development. Dev Biol 166:170–179.
Looijenga L, Gillis AJM, Verkerk AJMH, Van Putten WLJ, and Oosterhuis JW (1999). Heterogeneous X inactivation in trophoblastic cells of human full-term female placentas. Am J Hum Genet 64:1445–1452.
McGrath JS and Solter D (1984). Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 37:179–183.
Mutter G, Chaponot ML, and Fletcher JA (1995). A polymerase chain reaction assay for non-random X chromosome inactivation identifies monoclonal endometrial cancers and precancers. Am J Pathol 146:501–508.
Pulford D, Falls JG, Killian JK, and Jirtle RL (1999). Polymorphisms, genomic imprinting and cancer susceptibility. Mutat Res 436:59–67.
Rachmilewitz J, Elkin M, Rosensaft J, Gelman-Kohan Z, Ariel I, Lustig O, Schneider T, Goshen R, Biran H, de Groot N, and Hochberg A (1995). H19 expression and tumorigenicity of choriocarcinoma derived cell lines. Oncogene 11:863–870.
Redolfi E, Pizzuti A, Di Bacco A, Susani L, Labella T, Affer M, Montagna C, Reinbold R, Mumm S, Vezzoni P, and Zucchi I (1999). Mapping of the MYCL2 processed gene to Xq22–23 and identification of an additional L MYC-related sequence in Xq27.2. FEBS Lett 446:273–277.
Ropers J, Wolff F, and Hitzeroth JW (1978). Preferential X inactivation in human placenta membranes: Is the paternal X inactive in early embryonic development of female mammals? Hum Genet 43:265–273.
Szulman A (1984). Syndromes of hydatidiform moles, partial VS complete. J Reprod Med 29:788–791.
Takagi J and Sasaki M (1975). Preferential inactivation of the paternally derived X chromosome in the extraembryonic membranes of the mouse. Nature 256:640–642.
Walsh CM, Miller SJ, Flam F, Fisher RA, and Ohlsson R (1995). Paternally derived H19 is differentially expressed malignant and nonmalignant trophoblast. Cancer Res 55:1111–1116.
Young RH, Kurman RJ, and Scully RE (1988). Proliferations and tumors of intermediate trophoblast of the placental site. Semin Diagn Pathol 5:223–237.
Acknowledgements
We are indebted to Dr. Jon S. Morrow, M.D., Ph.D. for his encouragement and critical review of the manuscript, and to Dr. Carlo B. Bifulco, M.D. for his generous help with the study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hui, P., Parkash, V., Perkins, A. et al. Pathogenesis of Placental Site Trophoblastic Tumor May Require the Presence of a Paternally Derived X Chromosome. Lab Invest 80, 965–972 (2000). https://doi.org/10.1038/labinvest.3780099
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780099
This article is cited by
-
Historical, morphological and clinical overview of placental site trophoblastic tumors: from bench to bedside
Archives of Gynecology and Obstetrics (2017)
