
Abstract
The relationship between bile duct damage and portal fibrosis in chronic liver diseases remains unclear. This study was designed to show whether human intrahepatic biliary epithelial cells can undergo epithelial–mesenchymal cell transition, thereby directly contributing to fibrogenesis. Primary human cholangiocytes were stimulated with transforming growth factor-β (TGFβ) or TGFβ-presenting T cells and examined for evidence of transition to a mesenchymal phenotype. Liver sections were labelled to detect antigens associated with biliary epithelial cells (cytokeratin 7 and 19 and E-cadherin), T cells (CD8), epithelial–mesenchymal transition (S100A4, vimentin and matrix metalloproteinase-2 (MMP-2)), myofibroblasts (α-smooth muscle actin) and intracellular signal-transduction mediated by phosphorylated (p)Smad 2/3; in situ hybridisation was performed to detect mRNA encoding TGFβ and S100A4. Stimulation of cultured cells with TGFβ induced the expression of pSmad2/3, S100A4 and α-smooth muscle actin; these cells became highly motile. Although normal bile ducts expressed ALK5 (TGFβ RI), low levels of TGFβ mRNA and nuclear pSmad2/3, they did not express S100A4, vimentin or MMP-2. However, TGFβ mRNA and nuclear pSmad2/3 were strongly expressed in damaged ducts, which also expressed S100A4, vimentin and MMP-2. Fibroblast-like cells which expressed S100A4 were present around many damaged bile ducts. Cells in the ‘ductular reaction’ expressed both epithelial and mesenchymal markers together with high levels of TGFβ mRNA and pSmad2/3. In conclusion, the cells forming small- and medium-sized bile ducts and the ductular reaction undergo EMT during chronic liver diseases, resulting in the formation of invasive fibroblasts; this process may be driven by a response to local TGFβ, possibly presented by infiltrating T cells.
Similar content being viewed by others


Main
Damage to the small- and medium-sized intra-hepatic bile ducts is a characteristic feature of a range of chronic inflammatory liver diseases including primary biliary cirrhosis (PBC), primary sclerosing cholangitis (PSC), ductopenic allograft rejection and graft-versus-host disease. Observations of at least partial resolution of the ‘vanishing bile duct syndrome’ (VBDS) following allograft rejection1 or transient exposure to drugs such as carbamazepine2, 3, 4 suggests that the loss of bile ducts and their constituent intrahepatic biliary epithelial cells (IBECs) might be reversible. Nevertheless, progressive cholestasis resulting from bile duct loss is conventionally thought to predispose to portal fibrosis and, ultimately, biliary cirrhosis. Furthermore, cholestasis is generally accompanied by an increase in bile ductules at the interface with the parenchyma—the so-called ductular reaction5, 6—which in turn is thought to be a driver for periportal fibrosis. The ductular reaction may also be observed in other end stage liver disease for example alcoholic cirrhosis where it may be responsible for perpetuation of fibrosis.7
IBECs are obvious targets for immune-mediated injury. There is a consensus that they can bind immune effector cells in vitro resulting in efficient cytolysis.8 This has led to development of a model in which VBDS is seen as the consequence of IBEC death, the void produced by epithelial loss becoming populated by activated fibroblasts. Recent studies suggest that a number of cell types within the liver can undergo transition to become myofibroblasts capable of matrix synthesis during fibrogenesis.9 These include activated fibroblasts, hepatic stellate cells, biliary epithelial cells and, most recently, hepatocytes.10, 11, 12 A potential contribution of extra-hepatic precursor cells has also been established by detection of bone marrow-derived myofibroblasts within the liver following non-sex-matched allogeneic bone marrow or liver transplantation.13
In damaged kidney, there is now convincing evidence that local epithelial–mesenchymal transition (EMT) is the predominant source of fibroblasts.14, 15 Unlike the cytolytic model of epithelial cell death followed by fibroblast repopulation, EMT can only occur if ductular epithelial cells survive to transform into invading fibroblasts following stimulation by micro-environmental factors such as transforming growth factor β (TGFβ).16 That is, cell plasticity protects from injury-induced cell death resulting in a heterogeneous fibroblast population.12
The proteomic features of EMT include loss of epithelial markers such as E-cadherin, Zo 1 and cytokeratins, and the acquisition of fibroblast markers including S100A4 (the human homologue of fibroblast-specific protein-1 (FSP-1)), vimentin, matrix metalloproteinases (MMP) 2 and 9, α-smooth muscle actin (αSMA), collagens, and TGFβ.15 Induction of S100A4 (FSP-1) occurs sufficiently early to visualise epithelial cells in situ during the initial phases of EMT.17, 18 This protein can modify cell motility and growth through interactions with the cytoskeleton19 and the C terminus of p53.20 In addition, induction of S100A4 may promote invasive properties by upregulation of MMP expression.21 S100A4 may play a central role during the induction and maintenance of the EMT phenotype.15
EMT has recently been shown to contribute to the pathology produced by bile duct ligation in a murine model, with hepatocyte growth factor ameliorating this process.22 While it has been reported that some early bile duct loss and portal fibrosis in human disease has a potential for beneficial reversibility,23 the mechanism for this process has not been determined. Demonstration of the contribution of EMT to the bile duct loss observed in chronic liver diseases would provide a new basis for understanding their pathogenesis and potential treatment.
In this study, primary IBECs were cultured with a range of stimuli including TGFβ1 and TGFβ-presenting T cells to establish whether these can induce EMT. Sections from normal liver and liver from patients with the archetypal bile duct-damaging diseases PBC and PSC and other chronic cholestatic disease were then examined by immunohistochemistry and in situ hybridisation to detect characteristic markers associated with EMT.
MATERIALS AND METHODS
Culture of Human Intrahepatic Biliary Epithelial Cells
Primary human IBECs were derived from digested liver tissue (ScienCell, CA, USA). Non-epithelial cells were removed from initial cultures by differential detachment followed by specific antibody panning to deplete endothelial cells; purified cells were validated as >95% IBECs on the basis of cytokeratin-19 expression (details from techsupport@sciencellonline.com). These cells were cultured in 25 cm2 flasks for up to five population doublings in proprietary medium (ScienCell) supplemented with 2% fetal bovine serum; the TGFβ concentration in this medium was less than 3.5 pg/ml (ELISA; R&D Systems, UK). When the cells had reached 70% confluency they were seeded into 4-well chamber slides (Permanox Lab-Tek; Nunc, Denmark). After culture for 48 h to allow firm cell attachment, the medium in selected wells was supplemented with 1 or 10 ng/ml TGFβ1 (R&D Systems). Alternatively, MOLT-16 lymphoblastoid T cells (DSMZ ACC 29) were added to some chambers at a density of 8.3 × 104 cells/cm2. In all cases, the cultures were examined after incubation for 72 h using a Leica TCS SP2 UV laser-scanning confocal microscope (LSCM: Leica GMBH, Germany).
The expression of cell-surface TGFβ by MOLT-16 cells was validated in accordance with previously published methodology24 using a polyclonal antilatency-associated peptide (LAP) antibody (R&D Systems: 1/100); the presence of bioactive, cell-surface TGFβ was confirmed using the MFB-F11 cell line.25 The labelled cell suspensions were examined by flow cytometry (FACscan, Becton Dickinson, UK) using WinMDI analysis software; cytospin preparations were examined using a LSCM (Leica) under oil at × 63 magnification.
Immunofluorescence Analysis of IBEC
Cultured IBECs were washed and fixed in 4% phosphate-buffered paraformaldehyde (25 min at 20°C) and permeabilised with 1% Triton X-100 in PBS (15 min at 4°C). After washing with PBS, the cells were treated with 5% BSA (Sigma, UK) in PBS for 1 h before incubation with primary antibodies specific for cytokeratin 7 (CK-7; Dako, UK: 1/50), S100A4 (Dako: 1/100), α-smooth muscle actin (αSMA; Sigma, UK: 1/400) or pSmad2/3 (Santa Cruz Biotechnology, USA: 1/100) in 5% BSA overnight at 4°C; appropriate non-specific antibodies or normal rabbit IgG were used for control. The cells were then washed with 0.2% Tween 20 (Sigma) in PBS before addition of appropriate FITC-conjugated secondary antibody reagents for 1 h at RT in the dark. Following two further washes the chamber structures were detached and the slides were mounted in fluorescence mounting medium (Dako).
Immunofluorescence-labelled preparations were analysed using a Leica TCS SP2 UV LSCM. The LSCM was set up to image appropriate areas of fluorochrome-labelled cells (FITC excitation 488 nm, emission 510–535 nm) and a Z-series of optical sections was collected at 1 μm intervals. The threshold of positivity for each antibody was set using appropriately treated or untreated cells incubated with control antibody or rabbit IgG. Maximum projections of the collected Z-series were examined for localisation of the fluorophores.
Assessing the Invasive Potential of Cultured IBEC
Growth factor-reduced Matrigel invasion chambers (BioCoat: Becton Dickinson) with 8 μm pores were rehydrated with culture medium for 2 h in a humidified tissue culture incubator. The chambers were then seeded at a density of 4 × 104 cells/cm2 with either untreated IBEC or cells which had been stimulated with 10 ng/ml of TGFβ1 for 48 h. After incubation for 24 h the non-invading cells were removed by wiping the upper surface of the filter with a cotton bud. The membranes were then fixed and stained with Diff-Quick (Merck, UK), cut from the insert housing and mounted on slides in DPX (BDH, UK). Cells which had penetrated both the extracellular matrix and the membrane pores were identified by light microscopy and quantified by calculation of mean numbers of invasive cells per high power field.
Tissue
Archival formalin-fixed biopsy and liver explant tissue blocks were obtained from the Freeman Hospital, Newcastle upon Tyne and categorised as follows: PBC (2), PSC (5), end-stage alcoholic cirrhosis (2), hepatic steatosis (4), acute acetaminophen injury (1), normal donor liver sampled immediately prior to transplant (2) and a liver block containing both hepatocellular carcinoma and normal tissue. The use of human tissue for this research was approved by the local Research Ethics Committee.
In Situ Hybridisation
Paraffin embedded sections of explant PBC liver tissue were used to detect TGFβ and S100A4 by in situ hybridisation of specific mRNA sequences using FITC-hyperlabelled antisense oligonucleotide probes specific for S100A4 and TGFβ1 mRNA (GeneDetect; New Zealand): S100A4: 5′-TCCACCTCGTTGTCCCTGTTGCTGTCCAAGTTGCTCATCA-3′ TGFβ1: a mixture of 5′-GAAGTTGGCATGGTAGCCCTTGGGCTCGTG-3′ and 5′-ATGGTAGCCCTTGGGCTCGTGGATCCACTT-3′.
Briefly, sections were dewaxed, rehydrated and washed with diethylpyrocarbonate (DEPC)-treated water and DEPC-PBS. The sections were post-fixed with 4% paraformaldehyde in DEPC-PBS (30 min at 20°C) and endogenous peroxidase activity was quenched by incubation with 3% H2O2 for 10 min prior to permeabilisation in citrate buffer, pH6.0 (Vector Laboratories, UK) at 97°C for 30 min and, after cooling for 20 min at RT, 30 min in 0.3% Triton X-100/DEPC-PBS. The sections were then overlaid with prehybridisation buffer (Dako) at 37°C (S100A4) or 32°C (TGFβ) for 2 h before washing with DEPC-PBS and addition of the antisense probe in prehybridisation buffer (1:100). Hybridisation occurred at 37°C (S100A4) or 32°C (TGFβ) for 22 h in a humid chamber followed by stringent washes in decreasing concentrations of saline–sodium citrate. Non-specific binding sites were blocked by incubation with antibody diluent (Dako) for 30 min before incubation with peroxidase-conjugated anti-FITC (Dako) at 1:100 in antibody diluent for 1 h. The probe was detected using the CSAII Biotin-free TSA System from Dako (DAB substrate) with TBS-Tween washes to minimise background. Slides were counterstained with Mayer's Haematoxylin and washed with distilled water, dehydrated and mounted in DPX. Control sections were incubated with sense oligonucleotide probes or hybridisation buffer only (no probe) for 22 h at 37 or 32°C before detection. All sections were analysed using a Leica DMR microscope (Leica) with image capture and Leica QWin software.
Antigen Detection in Tissue Sections
All protocols were based on those previously described for kidney.18 Heat-induced epitope retrieval in citrate buffer was followed by an endogenous biotin blocking procedure (Vector) and incubation with non-immune animal serum. Primary antibodies were added at appropriate dilutions, overnight at 4°C. For single-labelling procedures the sections were incubated with either rabbit antihuman S100A4 (1/400), anti-MMP-2 (Serotec, UK: 1/100), anti-pSmad2/3 (1/400), anti-TGFβ RI (ALK5: Santa Cruz, USA), monoclonal mouse antihuman CK-7 or CK-19 (Dako: 1/50), E-cadherin (Dako 1/25), vimentin (Dako: 1/50) or anti-αSMA (1/4000). Alternatively, anti-S100A4 and mouse antihuman CD8, CK-19 or E-cadherin primary antibodies were applied simultaneously when dual-labelling was required. Negative control sections were incubated with appropriate control immunoglobulins in place of the primary antibodies. The secondary antibodies (1 h at RT) were biotinylated swine antirabbit immunoglobulins (Dako: 1/250) or rabbit antimouse immunoglobulins (Dako: 1/250).
Following single-antigen labelling, bound antibodies were visualised using either a streptavidin–biotin–peroxidase complex (SABPx; Vector) with nickel DAB (black; Vector) as chromagen or avidin–biotin–alkaline phosphatase complex (ABAP; Vector) and Vector Red chromagen. For dual-antigen labelling, the detection of individual bound primary antibodies was carried out sequentially. CD8, E-cadherin or pSmad2/3 were visualised first with SABPx and nickel DAB followed by visualisation of S100A4 with ABAP or AP-labelled Envision polymer (Dako) and Vector Red. Dual labelling of tissue sections to detect CK-19 and S100A4 was performed using an EnVision™ G∣2 Doublestain System, Rabbit/Mouse (DAB/Permanent Red) kit (Dako) according to manufacturer's instructions. Immunolabelled sections were analysed using a Leica DMR microscope (Leica, UK) with image capture and Leica QWin software.
For two-colour immunofluorescence labelling, heat-induced epitope retrieval and primary antibody labelling procedures were as above and secondary labelling was with TRITC-labelled swine antirabbit immunoglobulins (Dako; 1/40) for S100A4 detection or FITC-labelled rabbit antimouse immunoglobulins (Dako; F(ab')2 fragments: 1/150) for CK-7 or CK-19 detection. Secondary antibodies were incubated simultaneously for 2 h at 20°C then mounted with antifade immunofluorescence mountant (Dako) or Vectashield hard-set mounting medium (Vector). Two-colour labelling to detect S100A4 and αSMA was carried out sequentially, detecting S100A4 first with TRITC-labelled swine antirabbit immunoglobulins (as above) followed by incubation of sections with anti-αSMA (as above) and detection with FITC-labelled rabbit antimouse immunoglobulins (1/150). To decrease background fluorescence in formalin-fixed tissue, the sections were incubated in 0.3% Sudan Black for 10 min at room temperature followed by four washes in TBS before mounting.
Immunofluorescence-labelled preparations were analysed using the Leica TCS SP2 UV LSCM. For two-colour labelled specimens, the LSCM was set up to image the fluorochromes sequentially to avoid possible ‘crosstalk’ between the two emission spectra (TRITC excitation 541 nm, emission 570–580 nm; FITC excitation 488 nm, emission 510–535 nm). The areas of interest were selected and projected Z-series were examined to localise the fluorochromes.
RESULTS
Immunofluorescence Analysis of Cultured IBEC
Resting IBEC showed an epithelioid cobblestone morphology (Figure 1a: upper panel) and more than 95% expressed CK-7 (Figure 1a: lower panel). However, matched cultures which had been stimulated for 72 h with TGFβ1 at 1 ng/ml (Figure 1b) and 10 ng/ml (Figure 1c: upper panel) underwent a phenotypic change to produce bipolar cells with a fibroblastic morphology and expression of CK-7 was lost (Figure 1c: lower panel); during this culture there was no cell detachment or obviously increased rate of cell death. Importantly, nuclear pSmad2/3 was absent from resting cells (Figure 1d) and no detectable accumulation of nuclear pSmad2/3 was observed after stimulation with TGFβ1 at 1 ng/ml (Figure 1e); however, stimulation for 72 h with TGFβ1 at 10 ng/ml caused a high level of accumulation of pSmad2/3 within the nucleus of each cell (Figure 1f). Resting IBECs showed little expression of either S100A4 (Figure 1g) or αSMA (Figure 1j) but stimulation for 72 h with TGFβ1 at 1 ng/ml increased the expression of both of these antigens (Figure 1h and k respectively); this increase was even more apparent after treatment with TGFβ1 at 10ng/ml (Figure 1i and l respectively), with αSMA showing a clear microfibrillar distribution within the cytoplasm of the IBEC.

Microscopic analysis of IBEC cultured in basal media or for 72 h in the presence of TGFβ1 at 1 ng/ml or 10 ng/ml. Resting cells showed a normal epithelioid morphology (a: upper panel) and expression of CK-7 (a: lower panel) with no detectable pSmad2/3 (d) and little expression of S100A4 (g) or αSMA (j). Stimulation with TGFβ1 caused a dramatic change in morphology with the cells adopting a uniform bipolar fibroblastic phenotype (b and c: upper panel) and loss of CK-7 (c: lower panel). The expression of pSmad2/3 was not affected by TGFβ1 at 1 ng/ml (e) but was markedly enhanced within the nuclei of cells exposed to TGFβ1 at 10 ng/ml (f). Treatment of IBEC with TGFβ1 also increased the expression of cytoplasmic S100A4 (h and i) and αSMA (k and l); in both cases a greater increase in antigen expression was produced by TGFβ1 at 10 ng/ml than at 1 ng/ml.
Induction of EMT by TGFβ-Presenting T Cells
The human lymphoblastoid T cell line MOLT-16 was shown to express cell-surface TGFβ by immunofluorescence detection of the TGFβ-associated LAP. The uniformity of this expression was demonstrated by both flow cytometry and fluorescence microscopy (Figure 2a); the biological activity of this TGFβ was confirmed using a sensitive reporter cell bioassay (data not shown). Resting monolayer IBEC cultures (Figure 2b) showed a marked increase in the expression of organised αSMA following the addition of MOLT-16 cells for 72 h (Figure 2c); the T cell-treated IBEC also showed a change in morphology similar to that observed following the addition of soluble TGFβ1.

Induction of αSMA in IBEC treated with TGFβ-presenting T cells. (a) Flow cytometry (grey line shows control results, the black line shows specific labelling) and immunofluorescence (immunofluorescence is shown in the left panel, bright field is shown in the right panel) show that the MOLT-16 T cell line expresses LAP, which is a surrogate marker for active TGFβ. (b) Resting IBEC express little αSMA, but (c) coculture with MOLT-16 cells for 72 h increases the expression of organised αSMA within elongated cells.
Invasive Capability of Stimulated IBEC
Measurement of the potential of IBEC to invade the artificial basement membrane, Matrigel showed that few resting cells could penetrate this matrix (Figure 3). However, stimulation of IBEC with TGFβ1 for 72 h before the assay generated cells with a highly invasive phenotype, as shown by the increased number of cells, which have penetrated the matrix and passed through the pores of the filter support (P<0.001; Figure 3). It was apparent that the TGFβ1-stimulated cells had adopted a bipolar phenotype while the few resting cells, which had invaded the matrix were more rounded.

Analysis of the invasion of resting (control) and TGFβ1-stimulated IBEC through Matrigel and an 8 μm-pore filter. Resting cells showed little potential for invasion, and the few migrant cells observed retained a rounded morphology. By contrast, cells which had been treated for 72 h with TGFβ1 efficiently invaded the artificial basement membrane (P<0.001) and showed a fibroblastoid, bipolar morphology.
In Situ Hybridisation in Tissue Sections and Nuclear Accumulation of pSmad2/3 Protein
An antisense oligonucleotide probe cocktail specific for mRNA encoding TGFβ showed a low level of hybridisation in the nuclei of bile duct cells in normal liver (Figure 4a). In addition, nuclear accumulation of pSmad2/3 was just detectable by immunohistochemistry (Figure 4d). However, there was no evidence of hybridisation of an antisense probe specific for S100A4 mRNA in normal liver (Figure 4g). Antisense probes for both TGFβ and S100A4 mRNA showed significant hybridisation in the nuclei of the majority of epithelial cells within small bile ducts in sections from an explanted PBC liver (Figure 4b and h); hybridisation was also observed within some adjacent interstitial cells and within the intraepithelial infiltrating cells. Low-level hybridisation was evident for TGFβ mRNA in the majority of hepatocytes and for S100A4 mRNA in hepatocytes at the interface with the portal tracts in PBC (not shown). The specificity of these reactions was verified in sequential sections by demonstration of minimal labelling by a sense sequence-specific probes (Figure 4c and i); this background was similar to that produced in the absence of any probe (not shown). Accumulation of pSmad2/3 protein was very strong in the nuclei of bile ducts in sections from explanted PBC (Figure 4e) and, significantly, bile ducts with strong nuclear accumulation of pSmad2/3 also expressed high levels of S100A4 protein (Figure 4f).

Demonstration of mRNA encoding TGFβ and S100A4 and accumulation of pSmad2/3 in bile ducts in normal and PBC sections. (a) A low expression of TGF-β1 mRNA was detected in small bile ducts in normal liver (brown) in comparison to (b) the strong signal from the TGF-β1 probe in biliary epithelium of PBC. (c) Specificity of the hybridisation was confirmed by a sense probe. (d) A low level of pSmad2/3 protein was detected in the nuclei of several epithelial cells (black) within small bile ducts in normal liver whereas in (e) intense accumulation of pSmad2/3 was observed in small bile ducts in PBC. (g) Hybridisation with the S100A4-specific probe did not detect any S100A4 mRNA in normal liver but (h) a high level of S100A4 mRNA was observed in biliary epithelium in PBC. (i) An S100A4 sense probe proved the specificity of antisense binding. (f) Dual-labelling showed that nuclear pSmad2/3 (dark brown) was accompanied by expression of cytoplasmic S100A4 protein (red: no counterstain). (b) and (h) Both TGF-β1 and S100A4 antisense probes in PBC-detected specific mRNAs in some adjacent interstitial cells and infiltrating immune cells.
Analysis of Immunohistochemistry in Liver Sections
Sections of normal human liver showed clear, localised expression of CK-19 and CK-7, defining the epithelial cells lining the intrahepatic bile ducts (Figure 5a). Matched serial sections were stained to visualise S100A4, but showed no expression associated with either the epithelial cells or the surrounding tissues (Figure 5b), except for occasional immune cells. Normal bile ducts did not express αSMA, vimentin or MMP-2 (not shown).

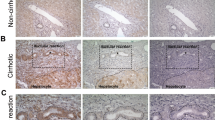
A representative series of immunohistochemical results from normal human liver and liver tissues from patients with PBC, PSC and alcoholic liver disease showing expression of S100A4, cytokeratins and E-cadherin. Serial sections (a and b) are from a normal liver biopsy taken immediately prior to transplantation; (a) shows expression of CK-19 in IBEC forming a small intrahepatic bile duct (arrow), (b) shows that neither this duct (arrow) nor the surrounding tissue expresses S100A4. (c) A dual-labelled section from stage 3 PSC detecting CK19 (brown/black) and S100A4 (red) shows a small bile duct (arrowed) in an inflamed portal tract, which is expressing S100A4 and a low level of CK19. (d) A section from Stage 3 PBC liver shows an S100A4-expressing epithelial cell (black arrow) within a ductule together with S100A4 expression within fibroblast-like cells in the surrounding tissue (white arrow). In PSC (Stage 4), (e) shows coexpression (yellow) of CK-19 (FITC-green) and S100A4 (TRITC-red) in a medium-sized bile duct and strong S100A4 expression (red) in infiltrating cells. (f) A large bile duct in stage 4 PSC showing coexpression (yellow) of CK-19 (FITC: green) and S100A4 (TRITC: red) in the majority of biliary epithelial cells (arrows identify strongly S100A4-positive epithelial cells). Most of the inflammatory cells surrounding the duct and infiltrating the epithelium are also S100A4-positive. (g) The damaged duct in Stage 4 PSC liver is surrounded by bipolar, S100A4-expressing cells (red) identified as invasive fibroblast-like cells, possibly resulting from localised EMT. (h and i) The arrows indicate ductular epithelial cells with very low expression of CK-19 (brown/black) that strongly express S100A4 (red) and appear to be acquiring a fibroblastic morphology (PSC stages 3 and 2 respectively). From Stage 3 PBC, (j) shows a medium-sized S100A4-expressing bile duct (red) infiltrated by CD8+ T cells (black: arrows). (k) Alcoholic cirrhosis; the upper panels show sequential confocal images identifying expression of CK-7 (green) and S100A4 (red) in epithelium in a ductular reaction. The cytofluorogram, derived from the same scan, shows evidence of extensive colocalisation of S100A4 and CK-7 in ductules (yellow pixels) with a band of red pixels on the left hand side representing intensely single-labelled infiltrating cells. The triple-labelled panel includes a DAPI nuclear counterstain with CK-7 and S100A4, again from the same scan; the narrow panel shows an XZ section through an individual ductule, confirming coexpression of CK-7 and S100A4 by single cells. (l) In stage 4, PSC a medium-sized bile duct shows fragmented and cytoplasmic E-cadherin expression and S100A4 in most of the epithelial cells. (m) Typical E-cadherin expression (black) in a section of normal bile duct.
Epithelial cells in bile ducts and ductules associated with the ductular reaction, in sections from PBC, PSC and ALD showed clear expression of S100A4 (Figure 5c–l). In addition, S100A4 and CK-19 (Figure 5c, e, f, h and i), S100A4 and CD8 (Figure 5j), and S100A4 and E-cadherin (Figure 5l) colocalised in bile ducts and in the ductular reaction within the diseased tissue. There was evidence of loss of CK-19 and E-cadherin and redistribution of E-cadherin to the cytoplasm in some S100A4-expressing epithelial cells and of a tendency to migrate out of the ductular structure, especially associated with the ductular reaction (Figure 5h and i).
Importantly, many cells within a ductular reaction (shown in alcoholic cirrhosis, Figure 5k) showed clear coexpression of both CK-7 and S100A4, defining a potentially motile population of cells with an epithelial phenotype undergoing localised EMT; the cells stained exclusively red are indicative of infiltrating leukocytes. An XZ optical section through a bile duct confirmed the coexpression of CK-7 and S100A4 by single epithelial cells (Figure 5k).
Epithelial cells in bile ducts and ductules associated with the ductular reaction, in sections from PBC, PSC and ALD also expressed vimentin (Figure 6a) and MMP-2 (Figure 6b); these cells did not express αSMA (Figure 6c and d).

A representative series of immunohistochemical results from normal human liver and liver tissues from patients with PBC, PSC and ALD showing expression of vimentin, MMP-2, αSMA, ALK5 and pSmad 2/3. (a) A vimentin expressing ductule (red) at the portal tract/hepatocyte interface (black arrow) lies in close proximity to vimentin-expressing fibroblast-like cells (grey arrow)—PBC Stage3. (b) The expression of MMP-2 (red) in a ductular reaction in Stage 4 PSC. (c) Shows the absence of αSMA (FITC: green) from epithelium within a small bile duct, which expresses S100A4 (TRITC: red); αSMA is present within myofibroblasts of the surrounding tissue (Stage 4 PSC). (d) Also in stage 4 PSC, a small bile duct confirms no coexpression of CK19 (TRITC: red) and αSMA (green: FITC-labelled primary antibody). By contrast the accompanying blood vessel is strongly αSMA-positive. (e) In a single-labelled section, a ductule in an infiltrated and fibrotic portal tract expresses S100A4, as do many of the infiltrating cells and hepatocytes (arrowed) at the interface with the parenchyma (end-stage ALD) (f) A single-labelled section from stage 3 PBC showing a bile duct-expressing ALK5 (TGFβRI—brown/black). Hepatocytes and a proportion of the infiltrating cells also express ALK5. (g) Coexpression of S100A4 and pSmad2/3; many of the S100A4 expressing cells (red) in a typical ductular reaction in end-stage alcoholic liver disease (ALD) also show nuclear localisation of the pSmad2/3 phosphoproteins (brown/black; no nuclear counterstain). (h) Negative method control—an ALD section in which primary antibody was replaced with rabbit IgG—counterstain, Mayer's haematoxylin.
S100A4 and vimentin were also observed within a population of infiltrating immune cells in the portal tracts. Some cells, identified morphologically as fibroblastic, expressed S100A4 (Figure 5d and g) and vimentin (Figure 6a); αSMA-expressing myofibroblasts were observed in areas of established fibrosis (Figure 6c and d). In addition, S100A4 (Figure 6e) and MMP-2 (not shown) were observed in hepatocytes adjacent to portal tracts.
ALK5 was strongly expressed in both native bile ducts and in the ductular reaction (Figure 6f) and expression of nuclear pSmad2/3 was increased in epithelial cells, when compared to normal (not shown), suggesting an increased response to active TGFβ; importantly, this signalling was spatially associated with the expression of S100A4 (shown in alcoholic cirrhosis; Figure 6g).
The small bile ducts in sections with hepatic steatosis expressed low levels of S100A4 (not shown) indicating a response to non-specific damage, but these biopsy sections did not contain medium-sized bile ducts or areas of ductular reaction. The bile ducts in sections from patients with acute liver damage were negative for both S100A4 and vimentin (not shown).
DISCUSSION
In this study, we have combined data from immunohistochemical and in situ hybridisation studies of human liver tissue with results from cell culture experiments to provide strong evidence that biliary epithelial cells contribute significantly to portal tract fibrosis in a number of cholangiopathies.
In a resting state, cultured IBEC had an epithelioid morphology and expressed both CK-7 and CK-19, but neither S100A4 nor αSMA; our group has shown that cytokeratin expression remains stable in these cells for more than seven passes in basal culture.26 Treatment with TGFβ1-induced Smad 2/3 phosphorylation with translocation of these phosphoproteins to the nucleus. Importantly, the stimulated cells uniformly adopted a fibroblastic morphology with increased cytoplasmic expression of the EMT marker, S100A4. They also acquired high levels of organised αSMA within their cytoplasm indicating transdifferentiation from epithelial cells to mature myofibroblasts, which had a significantly enhanced potential to invade a basement membrane-like matrix.
Neither IBEC nor other cells (except for occasional inflammatory cells) in the normal portal tract expressed S100A4, suggesting that any resident fibroblasts are of a lineage separate from those derived from local epithelial cells. It has already been reported that primary mesenchymal cells do not express S100A4.27 However, IBEC and cells with an epithelial phenotype within the ductular reaction from patients with PBC, PSC and alcoholic cirrhosis showed marked induction of S100A4, MMP-2 and vimentin; the induction of S100A4 was confirmed at an mRNA level. In addition, reduced expression of both cytokeratins and E-cadherin was noted in bile duct epithelium undergoing EMT. E-cadherin that remained was fragmented and was redistributed to the cytoplasm. This suggests that, at least in these diseases, the development of portal tract fibrosis is associated with local induction of an EMT process. Importantly, IBEC in biopsy sections did not express αSMA, but this antigen was expressed by myofibroblasts in established portal fibrosis, further emphasising the importance of S100A4 as an early marker of EMT.
The apparent disparity between the expression of αSMA by TGFβ-stimulated IBEC in culture and the absence of this antigen from epithelial cells which constitute recognisable bile ducts in diseased liver tissue can be explained by differences between the two systems. In both cases, individual epithelial cells clearly acquire some mesenchymal antigens and lose epithelial markers. This change is accompanied by an increase in motility and matrix invasion in vitro, which is entirely consistent with the observed induction of MMP-2 expression in vivo. Demonstration of increased S100A4 expression together with low levels of CK-19 in some cells with a fibroblastic morphology also suggests the presence of some cells at an early stage after the induction of EMT. Hence, the absence of αSMA within the ductular epithelium might simply reflect the fact that cells undergoing EMT have migrated from this tissue before acquiring αSMA expression. Kinetic differences in the expression of individual antigens after the induction of EMT further emphasises the importance of the detection within epithelial cells of S100A4, which has already been described as one of the earliest markers of this transition process.17 Indeed, S100A4 directly increases cell motility through modification of actin function, suggesting that expression of this marker is not compatible with long-term cellular localisation within organised epithelial tissues.28
As S100A4 expression by macrophages within the infiltrating leukocyte population may be confused with that of adjacent epithelial cells entering EMT,29 it is important to co-label with markers of differentiated epithelium such as E-cadherin and cytokeratins. Coexpression of CK-7 or CK-19 and S100A4 within small bile ducts and the majority of cells comprising the ductular reaction provided clear evidence that although these cells have an epithelial phenotype, they are actively engaged in EMT. This provides a possible explanation for the association between the ductular reaction and development of periportal fibrosis.30 The ductular reaction is seen in a number of end stage liver diseases, including alcoholic cirrhosis. It is particularly seen in areas of parenchymal extinction and may be responsible for progressive fibrosis in advanced disease. Expression of S100A4 and MMP-2 by some peri-portal hepatocytes is consistent with a previous study showing that these cells are also capable of undergoing EMT in vitro.12, 31 It is significant that CD8+ T-cells were in close contact with viable, S100A4-expressing IBEC in PBC, suggesting the potential for sophisticated crosstalk in the absence of target cell lysis. Such a scenario is clearly important for immune cell-mediated induction of localised EMT.18
Identification of coexpression of S100A4 and phosphorylated forms of Smad 2/3 in the nucleus of ductular epithelial cells provides evidence that this EMT process, as in renal fibrosis,32, 33 is driven by microenvironmental TGFβ. This is supported by the demonstration that nuclear expression of pSmad2/3 by cultured IBEC increased rapidly following stimulation with TGFβ1. A potential role for TGFβ as an effector molecule in PBC is also supported by the demonstration of increased TGFβ mRNA, confirming previous reports.34
An attractive candidate for TGFβ presentation to epithelial cells during persistent inflammation is T cells, which express the αE(CD103)β7-integrin,35 allowing adhesion to epithelial E-cadherin.36 In this study, a TGFβ-presenting, CD103+ T cell line induced EMT within cultured IBEC; similar results have been reported after culture of renal tubular epithelial cells with these T cells.18 Demonstration of the presence of LAP on the surface of CD103+ T cells provides a surrogate marker for the presence of active TGFβ, as the cells also express thrombospondin, which can activate the latent protein complex.24 Increased numbers of CD103+ve T cells have been reported within the portal infiltrate during PBC37 and chronic hepatitis.38
An important feature of EMT is its potential for reversibility. In tubulointerstitial renal fibrosis, tubular epithelial cells acquire S100A4 and the capacity to penetrate the basement membrane;39 with continued stimulation, some of these cells can differentiate further into proliferating myofibroblasts. Kalluri and Neilson15 have argued that the phenotype of these cells may remain plastic allowing phenotypic reversion of at least some cells and the reformation of functional tubular epithelium. Our data suggest a similar process may occur in liver disease since we have shown that IBEC have the potential to acquire an immature epithelial cell/fibroblast phenotype and progress to αSMA-expressing myofibroblasts. It is tempting to speculate that these cells retain their plasticity and, given the right conditions, could revert to form normal bile ducts. Such a scenario might explain the reversibility of vanishing bile duct syndrome in some situations.23
References
Hubscher SG, Buckels JA, Elias E, et al. Vanishing bile-duct syndrome following liver transplantation—is it reversible? Transplantation 1991;51:1004–1010.
Yao F, Behling CA, Saab S, et al. Trimethoprim–sulfamethoxazole-induced vanishing bile duct syndrome. Am J Gastroenterol 1997;92:167–169.
Ramos AM, Gayotto LC, Clemete CM, et al. Reversible vanishing bile duct syndrome induced by carbamazepine. Eur J Gastroenterol Hepatol 2002;14:1019–1022.
Taghian M, Tran TA, Bresson-Hadni S, et al. Acute vanishing bile duct syndrome after ibuprofen therapy in a child. J Paed 2004;145:273–276.
Burt AD, MacSween RN . Bile duct proliferation—its true significance? Histopathology 1993;23:599–602.
Roskams T, Desmet V . Ductular reaction and its diagnostic significance. Semin Diagn Pathol 1998;15:259–269.
Yip WW, Burt AD . Alcoholic liver disease. Semin Diagn Pathol 2006;23:149–160.
Leon MP, Bassendine MF, Gibbs P, et al. Immunogenicity of biliary epithelium: study of the adhesive interaction with lymphocytes. Gastroenterology 1997;112:968–977.
Ramadori G, Saile B . Portal tract fibrogenesis in the liver. Lab Invest 2004;84:153–159.
Robertson H, Kirby JA, Yip WW, et al. Biliary epithelial cell to mesenchymal cell transition in post-transplant recurrence of primary biliary cirrhosis. Hepatology 2007;45:977–981.
Sicklick JK, Choi SS, Bustamante M, et al. Evidence for epithelial–mesenchymal transitions in adult liver cells. Am J Physiol Gastrointest Liver Physiol 2006;291:G575–G583.
Zeisberg M, Yang C, Martino M, et al. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem 2007;282:23337–23347.
Forbes SJ, Russo FP, Rey V, et al. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology 2004;126:955–963.
Iwano M, Plieth D, Danoff TM . Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002;110:341–350.
Kalluri R, Neilson EG . Epithelial–mesenchymal transition and its implications for fibrosis. J Clin Invest 2003;112:1776–1784.
Liu Y . Epithelial to mesenchymal transition in renal fibrogenesis: pathologic significance, molecular mechanism, and therapeutic intervention. J Am Soc Nephrol 2004;15:1–12.
Okada H, Danoff TM, Kalluri R, et al. Early role of Fsp1 in epithelial-mesenchymal transformation. Am J Physiol 1997;273 (Part 2):F563–F574.
Robertson H, Ali S, McDonnell BJ, et al. Chronic renal allograft dysfunction: the role of T cell-mediated tubular epithelial to mesenchymal cell transition. J Am Soc Nephrol 2004;15:390–397.
Barraclough R . Calcium-binding protein S100A4 in health and disease. Biochim Biophys Acta 1998;1448:190–199.
Grigorian M, Andresen S, Tulchinsky E, et al. Tumor suppressor p53 protein is a new target for the metastasis-associated Mts1/S100A4 protein: functional consequences of their interaction. J Biol Chem 2001;276:22699–22708.
Bjornland K, Winberg JO, Odegaard OT, et al. S100A4 involvement in metastasis: deregulation of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases in osteosarcoma cells transfected with an anti-S100A4 ribozyme. Cancer Res 1999;59:4702–4708.
Xia JL, Dai C, Michalopoulos GK, et al. Hepatocyte growth factor attenuates liver fibrosis induced by bile duct ligation. Am J Pathol 2006;168:1500–1512.
Blakolmer K, Seaberg EC, Batts K, et al. Analysis of the reversibility of chronic liver allograft rejection implications for a staging schema. Am J Surg Pathol 1999;23:1328–1339.
Oida T, Zhang X, Goto M, et al. CD4+CD25- T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. J Immunol 2003;170:2516–2522.
Tesseur I, Zou K, Berber E, et al. Highly sensitive and specific bioassay for measuring bioactive TGF-β. BMC cell biology 2006;7:15.
Leon MP, Kirby JA, Gibbs P, et al. Immunogenicity of biliary epithelial cells: study of the expression of B7 molecules. J Hepatol 1995;22:591–595.
Strutz F, Okada H, Lo CW, et al. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol 1995;130:393–405.
Venkov CD, Link AJ, Jennings JL, et al. A proximal activator of transcription in epithelial–mesenchymal transition. J Clin Invest 2007;117:482–491.
Inoue T, Plieth D, Venkov CD, et al. Antibodies against macrophages that overlap in specificity with fibroblasts. Kidney Int 2005;67:2488–2493.
Ray MB, Mendenhall CL, French SW, et al. Bile duct changes in alcoholic liver disease. The Veterans Administration Cooperative Study Group. Liver 1993;13:36–45.
Valdes F, Alvarez AM, Locascio A, et al. The epithelial mesenchymal transition confers resistance to the apoptotic effects of transforming growth factor β in fetal rat hepatocytes. Mol Cancer Res 2002;1:68–78.
Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 2003;9:964–968.
Tyler JR, Robertson H, Booth TA, et al. Chronic allograft nephropathy: intraepithelial signals generated by transforming growth factor-β and bone morphogenetic protein-7. Am J Transplant 2006;6:1367–1376.
Harada K, Van de Water J, Leung PS, et al. In situ nucleic acid hybridisation of cytokines in primary biliary cirrhosis: predominance of the Th1 subset. Hepatology 1997;25:791–796.
Lehmann J, Huehn J, de la Rosa M, et al. Expression of the integrin alpha E β 7 identifies unique subsets of CD25+ as well as CD25- regulatory T cells. Proc Natl Acad Sci USA 2002;99:13031–13036.
Cepek KL, Shaw SK, Parker CM, et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E β 7 integrin. Nature 1994;372:190–193.
Yeaman SJ, Kirby JA, Jones DEJ . Autoreactive responses to pyruvate dehydrogenase complex in the pathogenesis of primary biliary cirrhosis. Immunol Rev 2000;174:238–249.
Shimizu Y, Minemura M, Murata H, et al. Preferential accumulation of CD103+ T cells in human livers; its association with extrathymic T cells. J Hepatol 2003;39:918–924.
Zeisberg M, Maeshima Y, Mosterman B, et al. Renal fibrosis. Extracellular matrix microenvironment regulates migratory behavior of activated tubular epithelial cells. Am J Pathol 2002;160:2001–2008.
Acknowledgements
Financial support: Roche Organ Transplantation Research Foundation No. 629106771. Marie Curie FP6 EU Programme No. 513904. Newcastle Healthcare Charity (RVI/NGH) and Newcastle upon Tyne Hospitals NHS Charity (FH).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rygiel, K., Robertson, H., Marshall, H. et al. Epithelial–mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab Invest 88, 112–123 (2008). https://doi.org/10.1038/labinvest.3700704
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700704
Keywords
This article is cited by
-
The crosstalk between cholangiocytes and hepatic stellate cells promotes the progression of epithelial-mesenchymal transition and periductal fibrosis during Clonorchis sinensis infection
Parasites & Vectors (2024)
-
A critical role of autophagy in regulating the mesenchymal transition of ductular cells in liver cirrhosis
Scientific Reports (2019)
-
Upregulation of NDRG1 predicts poor outcome and facilitates disease progression by influencing the EMT process in bladder cancer
Scientific Reports (2019)
-
2-Methoxyestradiol attenuates liver fibrosis in mice: implications for M2 macrophages
Naunyn-Schmiedeberg's Archives of Pharmacology (2019)
-
The role of the epithelial-to-mesenchymal transition (EMT) in diseases of the salivary glands
Histochemistry and Cell Biology (2018)
