
Abstract
Expression of the DNA repair protein O6-alkylguanine-DNA-alkyltransferase (AGT), encoded by the O6-methylguanine (O6-mG) -DNA-methyltransferase (MGMT) DNA repair gene, results in resistance to alkylating agents, and hypermethylation of the MGMT promoter is associated with chemosensitivity as it prevents AGT expression. As the interpretation of the results of immunohistochemistry to evaluate AGT expression proved to be difficult, the aim of our present study is to establish a feasible, reliable, and robust method for MGMT promoter hypermethylation testing that can be easily implemented in a diagnostic setting and is applicable to routinely processed tissue. MGMT hypermethylation analysis using methylation-specific (MS-) multiplex ligation-dependent probe amplification (MLPA) was performed on 62 glioma samples of 55 individual tumors (including 12 cell lines) and compared to the more conventionally used, but improved, MS-polymerase chain reaction (PCR). In contrast to MS-PCR, MS-MLPA (i) is not based on bisulfite conversion of unmethylated cytosines (a somewhat troublesome step in MS-PCR), (ii) provided methylation status of all samples, (iii) proved to be semiquantitative, (iv) can be used to evaluate methylation status of multiple sequences (CpG dinucleotides) simultaneously, and (v) allows for a combined copy number detection and methylation specific analysis. The potential therapeutic value of MGMT hypermethylation evaluation using MS-MLPA was shown in a group of 20 glioblastoma patients receiving temozolomide chemotherapy. We conclude that MS-MLPA is a robust and reliable method that can be easily applied to differently processed tissues, including those fixed in formalin and embedded in paraffin. The semiquantitative aspect of MS-MLPA may prove to be of great value, especially in predicting response to alkylating agents, not only for gliomas as evaluated in this study but also for tumors in general.
Similar content being viewed by others


Main
O6-alkylguanine-DNA-alkyltransferase (AGT) is an important DNA-repair protein encoded by the gene O6-methylguanine-DNA-methyltransferase (MGMT) (reviewed by Drablos et al;1 Esteller and Herman;2 Gerson3). Scanning DNA by flipping each base into the active-site pocket, AGT removes alkyl adducts at the O6 position of guanine and, to a lesser extent, at the O4 position of thymine. After repair, the alkylated MGMT protein is released and targeted for degradation by ubiquitination.4 Repair of these DNA lesions is essential for cell integrity, and absence of expression of the MGMT gene is a poor prognostic factor for several cancers as it leads to accumulation of mutations (eg, O6-methyl-guanine mispairs with thymine) and chromosome instability.5 During chemotherapy with alkylating agents, however, one of the major mechanisms inducing resistance is through the demethylating activity of AGT removing the added alkyl adducts.6
Silencing of the MGMT gene has been associated with MGMT promoter hypermethylation in a wide spectrum of tumors, including gliomas.2 Tumors with a hypermethylated MGMT promoter were reported to be more sensitive to alkylating agents, such as carmustine (BCNU), procarbazine, lomustine (CCNU), and temozolomide (TMZ), compared to tumors without this MGMT silencing.7, 8 Aiming for tailor-made treatment, it is therefore important to identify whether MGMT is silenced in a specific tumor.
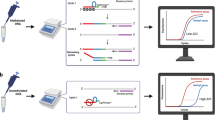
Currently, methods used to establish promoter hypermethylation are based on bisulfite conversion of normal (unmethylated) cytosine nucleotides into uracil, leaving methylated cytosine intact. As the specific methylation sites in the promoter are known, a subsequent polymerase chain reaction (PCR) can be performed using two sets of primers located in the hypermethylated region that specifically generate amplification products in either methylated DNA (M) or unmethylated DNA (U) (MS-PCR).9, 10 As multiple CpG dinucleotides are located in these primers, PCR product will only be generated when either all CpGs are M or U, whereas sequences with methylation of only part of these CpGs cannot be evaluated. Furthermore, bisulfite treatment is (for a variety of reasons) not always efficient,11, 12 which negatively affects the reliability of these assays, especially when using formalin fixed, paraffin-embedded tissue. Although the development of new bisulfite conversion kits significantly improved the conversion rates, other robust approaches remain warranted to detect these prognostically and therapeutically important aberrations. Recently, the application of methylation-specific (MS-) multiplex ligation-dependent probe amplification (MLPA) (outlined in Figure 1) was described as a method to evaluate the methylation status of multiple CpG dinucleotides individually.13 MS-MLPA has the advantage that bisulfite conversion can be omitted, different CpG dinucleotides can be analyzed simultaneously, low-molecular-weight DNA isolated from paraffin-embedded tissue (p-DNA) can be analyzed as the MLPA probes are small (approximately 50–60 bp), only a small amount of DNA is required (100 ng), and copy number detection of the analyzed loci can be performed simultaneously.

Schematic outline of the procedure for methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA). (a) Ordinary MLPA probes harbor two oligonucleotides, one short synthetic and one long M13-derived oligonucleotide. Up to 45 probes can be added to each MLPA reaction. Both oligonucleotides contain universal primers sites and the M13-derived oligonucleotide contains a stuffer sequence that varies in length between the different probes. For MS-MLPA, the probe design is similar to an ordinary MLPA probe, except that, for the methylation-specific probes, the sequences detected contain a methylation-sensitive restriction site (HhaI; indicated as a blocked box in the target specific sequence). To establish the amount of methylated sequences after hybridization of the probe mix to the sample DNA, the sample is split and one part is subjected to a single ligation step, whereas for the other part ligation is combined with the methylation-sensitive digestion. Subsequent PCR amplification therefore exponentially amplifies either total DNA or the methylated fraction only. Comparing the peak-signals of the ligated fraction and the fraction that is also digested with HhaI generates the methylation ratios. Furthermore, by comparing peak-signals of the ligated fraction with undigested control DNA evaluated in parallel will provide information about the copy number of the evaluated probes. (b) Capillary gel electrophoresis is performed to identify and quantify PCR products of the individual probes. Methylation-sensitive probes are indicated in boxes and show MGMT1 (3rd box), MGMT2 (5th box) and MGMT3 (6th box). The undigested part shows the same amount of PCR product as the sample containing 100% (in vitro) methylated DNA, whereas peak height for the probes containing a MS-sensitive digestion site gradually decreases as the amount of methylated sequences decreases.
In the present study, we first illustrate that interpretation of the results of immunohistochemistry to evaluate MGMT protein expression can be difficult. Consequently, there is a need for a feasible, reliable method for MGMT promoter hypermethylation testing. The main goal of this study is to validate detection of MGMT hypermethylation using MS-MLPA in 62 samples of 55 individual gliomas (including 12 cell lines and 43 biopsies) and to compare these results to the more conventional MS-PCR. Furthermore, we established the occurrence of MGMT promoter hypermethylation, MGMT copy number losses, and its clinical relevance in a subset of glioblastoma multiforme (GBM) patients treated with TMZ.
Materials and methods
Tumor Samples
For this study, samples were retrieved from our neuro-oncology archive at the Department of Pathology of the Radboud University Nijmegen Medical Centre and the Canisius Wilhelmina Hospital in Nijmegen, The Netherlands. The use of brain tumor tissue after completing histopathological diagnosis for research purposes has been approved by the regional ethics committee. Tumors were classified and graded according to the WHO-2000 classification as diffuse astrocytoma (A-II), anaplastic astrocytoma (A-III), GBM, oligodendroglioma (O-II), anaplastic oligodendroglioma (O-III), oligoastrocytoma (OA-II), and anaplastic oligoastrocytoma (OA-III).14
Thirty-three glioma biopsies were subjected to immunohistochemical analysis of MGMT/AGT including: 2 A-IIs, 1 A-III, 14 GBMs, 2 O-IIs, 5 O-IIIs, 4 OA-IIs, and 5 OA-IIIs. For hypermethylation analysis, 12 glioma cell lines, 50 samples of 43 gliomas (1 A-II, 4 A-IIIs, 29 GBMs, 2 O-IIs, 4 O-IIIs, and 3 OA-IIIs), and one sample of normal brain were evaluated.
Immunohistochemistry
Immunohistochemical analysis for MGMT was performed using formalin-fixed, paraffin-embedded tissue and a commercially available, monoclonal anti-MGMT antibody (Chemicon, Hampshire S053 4NF, UK). The staining procedure was performed as follows: after deparaffinizing 4 μm sections in xylol (3 × 5 min), endogenous peroxidase was blocked by H2O2-PBS (10%). Antigen recovery was achieved by sodiumcitrate incubation in a microwave (3 min at 360 W followed by 10 min at 90 W). Sections were preincubated with normal horse serum (20%) in PBS-BSA (20 min at room temperature), followed by incubation with the MGMT antibody (1/1600 in PBS-BSA). After removing aspecifically bound antibody, the sections were incubated with a secondary antibody (Dako Cytomation, Glostrup, Denmark), which was detected using the Vectastain avidine/biotine/peroxidase complex (Vectorlab, Burlingame, CA, USA) and visualized by AEC. Slides were counterstained with hematoxilin and mounted. Formalin-fixed, paraffin-embedded tonsil tissue was used as positive control.
DNA Isolation
DNA was isolated from routinely processed, formalin-fixed, paraffin-embedded materials (p-DNA) and from frozen tissue (f-DNA). In case of paraffin-embedded tissue, 50 μm paraffin sections were cut and incubated in P-buffer (50 mM Tris-HCl pH 8.2, 100 mM NaCl, 1 mM EDTA, 0.5% Tween-20, 0.5% NP40, 20 mM DTT) at 90°C for 15 min after which a protein digestion was performed using 0.5 mg/ml proteinase K (Roche Diagnostics GmbH, Mannheim, Germany) at 55°C overnight and another 48 h at 37°C (fresh proteinase K being added every 24 h). Subsequently, DNA was isolated using the DNeasy Tissue Kit (Qiagen, Venlo, The Netherlands). DNA was isolated from snap-frozen tumor tissue with this kit, as described by the manufacturer. Furthermore, DNA previously isolated using a salting out procedure15, 16 was purified using the DNeasy Tissue kit. An additional wash step using the AW2 wash buffer was always included before elution.
Methylation-Specific PCR: MS-PCR
Bisulfite conversion of unmethylated Cytosine was performed using the EZ DNA methylation-Gold kit as described by the manufacturer (Zymo Research, Orange, CA, USA). CT conversion reagent was prepared as described, and during incubation, the solution was protected from light. For every experiment, a fresh solution was prepared and used immediately. A volume of 130 μl was added to 20 μl DNA (1 μg) and protected from evaporation by mineral oil. Incubation was performed in a thermocycler, as recommended for longer DNA fragments: 10 min at 98°C and 30 min at 53°C, followed by 8 cycles of 6 min at 53°C and 30 min at 37°C, and a final hold at 4°C. Columns were prepared by adding 600 μl M-binding buffer, and the samples were loaded immediately after the incubations were finished. Sample and buffer were mixed before centrifugation and columns were washed using M-Wash Buffer. Columns were incubated with 200 μl of M-Desulfonation buffer (15 min at room temperature, protected from light) and washed twice with M-Wash buffer. Samples were eluted using 10 μl of elution buffer and immediately used for MS-PCR.
MS-PCR was performed as previously described with minor modifications.17 Briefly, PCR amplification was performed in a total volume of 25 μl using 2 μl of bisulfite-treated DNA, buffer IV (Integro, Dieren, The Netherlands), 200 mM of each deoxynucleotide triphospate, 1.0 or 1.5 mM MgCl2 (M and U primer set, respectively), 0.5 pmol of each primer, and 0.5 U of thermostable DNA polymerase (Integro, Dieren, The Netherlands). Primers used to detect unmethylated and methylated MGMT sequences, respectively, encompassed: U-MGMT-forward TTTGTGTTTTGATGTTTGTAGGTTTTTGT and U-MGMT-reverse AACTCCACACTCTTCCAAAAACAAAACA (93 bp), and M-MGMT-forward TTTCGACGTTCGTAGGTTTTCGC and M-MGMT-reverse GCACTCTTCCGAAAACGAAACG (81 bp).17 Recognition sites of the primers are indicated in Figure 2. First, DNA and MQ were mixed and a hotstart was performed for 5 min at 95°C to ensure complete denaturation of the GC-rich promoter region. After this, the other components were added and the following steps were performed: 35 cycles of denaturation for 60 s at 95°C, annealing for 50 s at 61°C, an extension step for 60 s at 72°C; a final extension for 7 min at 72°C; and a hold at 4°C. DNA amplification was performed in a PTC 200 thermal cycler (MJ Research Inc., Waltham, Massachusetts, USA). PCR amplification products (10 μl) were analyzed using agarose gel electrophoresis (3% gel to separate small products).

Location of the probes used to analyze MGMT hypermethylation. Genomic MGMT sequence according to the GenBank entry NT-008818. Sequences recognized by all four MS-MLPA probe sets (MGMT1, MGMT2, MGMT3, and MGMT4) are indicated in boxes. For each probe set, both individual parts are indicated as well as the recognition site for HhaI (GCGC, shown in bold). Both regions to which the MS-PCR primers hybridize after bisulfite treatment of the DNA are indicated in grey boxes and included CpG sites are underlined. The primers used to detect methylated sequences are indicated in dark grey and generate a 81-bp PCR fragment. The primers used to detect unmethylated sequences encompass the same regions but are six nucleotides longer each (indicated in light grey) and generate a 93-bp PCR fragment. Exon 1 is shown in italics.
Methylation-Specific Multiplex Ligation-Dependent Probe Amplification
MS-MLPA procedure
The MS-MLPA probe mix used was prepared by MRC-Holland (Amsterdam, The Netherlands) and contains 18 probe sequences of which 10 sequences (control probes) are not influenced by HhaI digestion (methylation sensitive restriction enzyme; recognition site GCGC). The eight probes used to detect hypermethylation contain an HhaI digestion site and encompass four MGMT probes: MGMT1 (5670-L5146; 193 bp); MGMT2 (2239-L1261; 373 bp); MGMT3 (5668-L5144; 454 bp); MGMT4 (5671-L5147; 319 bp). Recognition sites of these MGMT probes are shown in Figure 2. All MLPA probe pairs code for unique human single-copy DNA sequences and were designed and prepared as described by Schouten et al18 (http://www.mlpa.com).
MLPA was performed as described by the manufacturer with minor modifications. Briefly, 5 μl DNA (100–200 ng) was denatured and subsequently cooled down to 25°C. After adding the probe mix, the sample was denatured and the probes were allowed to hybridize (16 h at 60°C). Subsequently, half of the sample was ligated, whereas for the other part of the samples, ligation was combined with an HhaI digestion resulting in ligation of the methylated sequences only. PCR was performed as described by the manufacturer; however, double PCR amounts were used (50 μl PCR volume containing 10 μl of the ligation reaction), because in our experience, this results in a major improvement of (reliable) copy number analysis. Agarose gel electrophoresis was used to check MLPA efficiency.19 Subsequently, PCR reaction fragments were separated by capillary gel electrophoresis (ABI 3730, Applied Biosystems) and quantified using the Genemapper software (Applied Biosystems).
For copy number detection, at least four normal control DNA samples were included in each set of MLPA experiments next to the tumor samples for data processing. Reference DNAs were isolated from blood from healthy volunteers or isolated from normal (non-tumorous) paraffin-embedded samples and used as control samples analyzing f-DNA or p-DNA, respectively.20
MS-MLPA data processing
Data analysis was performed in Excel as described by the manufacturer of the MLPA kits. To compensate for differences in PCR efficiency of the individual samples, the fraction of each peak is calculated by dividing the peak value of each probe amplification product by the combined value of the control probes within the sample. For hypermethylation analysis, this ‘relative peak value’ or so-called ‘probe fraction’ of the digested sample is divided by the ‘relative peak value’ of the corresponding undigested sample generating the ‘methylation ratio’. From our dilution experiments, it became clear that the ratio indicates the percentage of methylated sequences. Duplicate experiments were performed for methylation analysis and average ratios were calculated.
Copy number analysis can be performed using the MLPA results of the undigested samples. After calculation of the ‘relative peak value’ or so-called ‘probe fraction’, this is divided by the ‘mean probe fraction’ of this fragment within the included reference DNAs, generating the ‘normalized peak value’ or the so-called ‘copy number ratio’. On the basis of our previous work using MLPA, thresholds to detect gains and losses were set at 1.2 and 0.8, respectively.19
Statistical Evaluation of MS-PCR and MS-MLPA
Cohen's Kappa analysis was used to assess the degree of agreement between MS-PCR and MS-MLPA. To compare the numerical MS-MLPA data with the categorical MS-PCR data, the MS-MLPA data were (in line with the clinically relevant MS-PCR categories) converted into two categories: hypermethylation being present (average probe ratio>0.25) or absent (average probe ratio <0.25). Performing statistical analysis, Kappa scores indicate poor (κ≤0.2), fair (0.2 <κ≤0.4), moderate (0.4 <κ≤0.6), substantial (0.6 <κ≤0.8), or good (κ>0.8) agreement.21
In Vitro Methylation Assay
To test the sensitivity of MS-PCR and MS-MLPA, a titration experiment was performed using normal DNA isolated from lymphocytes that were methylated in vitro using SSSI as described by the manufacturer (New England Biolabs, Ipswich, USA). Methylated samples were diluted to 75% methylated (M), 50% M, and 25% M using the original unmethylated DNA.
Results
Immunohistochemical Detection of MGMT Expression
Immunohistochemical analysis of the MGMT protein AGT was performed on 33 gliomas. Examples of immunohistochemical detection of AGT are shown in Figure 3 in combination with the available MS-MLPA data. In many cases, only part of the cells in the tumor biopsy showed nuclear staining, and there was a poor correlation between the immunohistochemical staining results and the hypermethylation status of the MGMT promoter as detected by MS-PCR or MS-MLPA. Occasionally, a clear-cut presence of AGT in the majority of the tumor cells was detected. As illustrated in Figure 3, the endothelial cells of the glioma vessels consistently showed immunohistochemical staining for MGMT and can therefore serve as an internal positive control confirming the quality of the staining procedure.

Examples of immunohistochemical staining for AGT (encoded by the gene MGMT) in gliomas showing different degrees of MGMT hypermethylation. AGT expression was always detected in the endothelial cells of the glioma microvasculature, these cells can therefore serve as a positive internal control (b and c). AGT expression of tumor cells was variable even when the MGMT hypermethylation status was taken in account. In N254 (a) extensive nuclear AGT expression was detected in the majority of cells, which correlates well with the absence of MGMT hypermethylation. However, in another tumor showing a comparable MGMT status (N448; b), nuclear expression of AGT was detected in only part of the cells. Similarly, different expression patterns were detected in tumors containing moderate MGMT promoter hypermethylation (c–f). In N41 (c) AGT could not be detected by immunohistochemistry, probably due to MGMT promoter hypermethylation. However, in N107 (d) showing a comparable degree of hypermethylation part of the cells do show AGT expression. The high number of AGT expressing cells in N293 (e) was rather unexpected because of the detection of hypermethylation. However, the MS-MLPA ratio of 0.58 in this tumor suggests that indeed part of the tumor cells may contain unmethylated MGMT allowing expression of AGT. Interestingly, elsewhere in the same tumor a clone of AGT expressing cells was present in an otherwise largely negative population of tumor cells (f), illustrating the heterogeneity of the tumor for AGT expression.
MGMT Hypermethylation: MS-PCR
Performing MS-PCR in combination with the newly developed EZ DNA Methylation-Gold kit (Zymo research), the majority of samples (56/58; 96%), even when using p-DNA (32/34; 94%), could be evaluated (Figure 4). For 45 cases, MS-PCR replicate experiments were performed (data not shown); however, MS-PCR did not generate results in all experiments (13/38 p-DNAs and 5/24 f-DNAs). Reliable results were available at least in duplicate for 36 cases, and although variability in the quantity of PCR amplification product was detected, this never affected the detection of methylated sequences. Overall, of the 12 GBM cell lines, M, MU (combination of methylated and unmethylated), or U were detected in 8, 2, and 2 of the cases, respectively, and in 7, 19, and 15, respectively, for the different tumors (2 cases provided no MS-PCR results).

MGMT analysis of gliomas. Results are shown for methylation detection by MS-PCR and MS-MLPA, and for MGMT copy number analysis as detected by MLPA, which was performed in parallel with MS-MLPA. Abbreviations used: World Health Organization (WHO) classification; GBM=glioblastoma multiform, A-II=diffuse astrocytoma, A-III=anaplastic astrocytoma, O-II=oligodendroglioma, O-III anaplastic oligodendroglioma, OA-III=anaplastic oligoastrocytoma, *=cell line samples. Copy number changes including the 10q26 MGMT region as detected by comparative genomic hybridization (CGH): −=loss (ratio <0.8); +=gain (ratio>1.2); n=normal (ratio in between 0.8 and 1.2); and when nothing is indicated samples were not subjected to CGH. For MS-PCR results are shown in gray or white boxes when a PCR amplification product was present or absent, respectively. Replicate experiments were performed, except for those indicated by a #. For some cases, not all replicate experiments provided MS-PCR results, this is shown on the right of the final result, e.g. 1/2=experiment was performed twice of which one did not generate product in both PCRs (U and M). For MLPA analysis, the different MGMT probes (1–3) are indicated as well as their average. For MS-MLPA analysis, duplicate experiments were performed, except for those indicated by a #, and the average of both is shown. To provide a better overview, the average methylation status is indicated in different shades of gray including light gray, gray and dark gray representing MS-ratio in between 0.25–0.5, in between 0.5–0.75, and above 0.75, respectively. For MLPA copy number detection, average ratios above 1.2 and below 0.8 are indicative for gains and losses, respectively, and are shown in green and red boxes respectively.
Using the same protocol required for reliable analysis of f-DNA and p-DNA, a titration experiment was performed analyzing samples containing 100, 75, 50, 25, and 0% in vitro methylated normal f-DNA (Figure 5). High quantities of PCR products were detected, but we could not distinguish between the 25–75% methylated samples, all showing U and M products. Only M or U were detected in the 100 and 0% methylated samples, respectively.

Titration experiment analyzing a dilution series of in vitro methylated DNA by MS-PCR or MS-MLPA. M represents methylated sequences whereas U represents unmethylated sequences. MS-MLPA; ratios are shown on the y-axis, whereas the three different MGMT probes, MGMT1, MGMT2, and MGMT3 are shown on the x-axis. Different amounts of methylated sequences are indicated as ♦=100% M, ▪=75% M, ▴=50% M, •=25%, and ★=0% M. Note that using the standard MS-PCR protocol allowing analysis of the majority of tumor samples including p-DNA, no distinction can be made between samples containing 25, 50, or 75% methylated sequences, whereas in the MS-MLPA analysis ratios decrease with decreasing amount of methylated sequences.
MGMT Hypermethylation: MS-MLPA
Reliability of the four MGMT MS-MLPA probes was first checked using normal unmethylated control f-DNA and p-DNA. As the MGMT4 probe was not digested in these control samples, this probe was excluded from further analysis. Probe MGMT2 gave ratios between 0.25 and 0.35 in four of the eight normal control p-DNAs, whereas hypermethylation of this probe was not detected in any of the eight control f-DNA. Using thresholds of 0.35 (instead of 0.25) for the methylation detection, when analyzing p-DNA, will allow reliable analysis of probe MGMT2.
A titration experiment was performed to determine whether the assay is semiquantitative (Figure 5). As illustrated in Figure 1, peak height of the methylation specific probes indeed gradually increased with the degree of methylation, which is reflected in the probe ratios shown in Figure 5. Therefore, methylation ratios are indicative of the amount of methylated DNA. Furthermore, using Cohen's Kappa analysis to assess the degree of agreement between MS-PCR and MS-MLPA in the detection of MGMT methylation, a good correlation was detected (κ=0.84).
MS-MLPA analysis was performed in duplicate for 57/62 samples analyzed producing reproducible ratios (data not shown). The average probe ratios of these duplicate experiments and the overall average of the three different CpG dinucleotides analyzed are shown in Figure 4. We interpreted (average) ratios as absence of hypermethylation (0.00–0.25), mild hypermethylation (0.25–0.50), moderate hypermethylation (0.50–0.75), and extensive hypermethylation (>0.75). Of the 12 cell-lines, only 2 showed absence of hypermethylation, whereas the others showed moderate (2 × ) or extensive (8 × ) hypermethylation. In the tumor biopsies, 16 × absence of, 15 × mild, 9 × moderate, and 3 × extensive hypermethylation were detected. For seven tumors, both p-DNA and f-DNA were evaluated and only once (N449) a minor difference was detected between both samples (f-DNA: 0.64 and p-DNA: 0.42), which might be attributed to tumor heterogeneity or tumor-load in the different specimens.
Correlation of MGMT Hypermethylation Status and Survival after TMZ Treatment
To get a first impression of the clinical relevance of MGMT MS-MLPA, the correlation between MS-ratios (overall average) and post operative survival of 20 patients who were treated with TMZ for GBM is shown in Figure 6. As expected, there is a trend toward better survival with increasing MS-MLPA ratios. To compare the predictive value of MS-PCR and MS-MLPA, cases in which methylated sequences were detected (M or UM) and those containing only unmethylated sequences were marked, and in general MS-PCR and MS-MLPA were comparable.

Survival analysis of 20 GBM patients in our study set receiving TMZ chemotherapy. Shown is the average MS-ratio for all three MGMT probes. Circles and diamonds indicate cases that, using MS-PCR, were identified as unmethylated or methylated (with or without additional unmethylated sequences), respectively. For the case indicated by an asterisk, no MS-PCR data could be generated. Open circles or diamonds represent patients that are still alive. Survival is shown in months. Note that there is a trend of a correlation between increasing ratios as detected by MS-MLPA and survival.
MGMT Copy Number Detection
All cell lines analyzed (n=12), and a small fraction of the tumor samples (n=5), were previously analyzed for (partial) chromosomal copy number changes, as detected by comparative genomic hybridization (CGH). All losses, including the MGMT gene located at 10q26 as detected by CGH, were also identified as a loss performing MLPA copy number analysis (showing an average ratio for the 3 MGMT probes below 0.8) (Figure 4). Of the nine cases, in which CGH did not identify copy number changes including 10q26, losses and gains were detected in two cases each, which is not unexpected, as CGH can only detect copy number changes >2 Mb, whereas MLPA has a much higher resolution.
Overall, MGMT copy number changes most frequently involved a loss (31/55 tumors), whereas a gain was detected in only five cases (9%). Excluding the GBM celllines, loss of MGMT was detected in 24 of the 43 tumors (70%), whereas a gain was detected in only 4 (11%). Differences between p-DNA and f-DNA (N293 and N452) might be caused by tumor heterogeneity (in the overview, the frozen sample was included). Distribution of the average MS-MLPA MGMT ratios is comparable for tumors harboring either an MGMT loss, gain, or have no MGMT copy number change. Loss of MGMT copy number did not correlate with chemoresponse (data not shown), and neither was a difference in survival detected between patients whose tumors showed hypermethylated sequences with or without copy number loss.
Discussion
MGMT is involved in DNA repair of alkyl adducts and plays a major role in resistance to chemotherapeutic agents, whereas loss of function may improve chemosensitivity. Identification of such chemosensitive tumors is important for tailor-made treatment: those patients who are most likely to be benefited from chemotherapy can be selected, whereas others are spared from the negative side effects and selected for alternative approaches without loss of time. Hypermethylation of the MGMT promoter was shown to be indicative of chemosensitivity. The currently used MS-PCR assays are based on the somewhat troublesome bisulfite treatment and two subsequent PCRs detecting either M or U sequences but not sequences with methylation of only part of the CpG dinucleotides.11, 12 Alternative and robust approaches for detection of the methylation status that are applicable in a routine diagnostic setting are therefore warranted.
Immunohistochemical Detection of MGMT Expression
Hypermethylation of the MGMT promoter inhibits expression of the protein. It has been reported that immunohistochemical detection or activity assays of the MGMT protein AGT can be used as tools to identify patients whose tumors are resistant to TMZ.6, 22 Immunohistochemistry has the advantage that it can be performed on archival, formalin-fixed, and paraffin-embedded tissue and may be helpful to sort out the contribution of ‘contaminating’ normal cells in the expression of MGMT. However, our results show that interpretation of the immunohistochemical staining results often remains difficult. Only a minority of the cases showed extensive AGT expression in the biopsy removed before chemotherapy, a finding that would predict chemoresistance to TMZ. In the majority of cases, however, only part of the cells in the biopsy specimen showed expression, and the following questions then need to be answered: (i) are the positive cells really tumor cells or non-neoplastic cells (eg, incorporated preexistent cells or inflammatory cells) expressing AGT? (ii) are the negative tumor cells incapable expressing AGT or can they be stimulated to produce AGT when subjected to a genotoxic drug? With regard to the first question, it is important to note that MGMT expression was reported to be present in non-neoplastic endothelial cells, astrocytes, oligodendroglial cells, and in inflammatory cells within a glioma.23 Furthermore, it has been demonstrated that MGMT expression can be induced by glucocorticoids, ionizing radiation, and genotoxic agents, including those used during chemotherapy.24, 25, 26 Determination of the hypermethylation status of the MGMT promoter therefore remains warranted, as it provides information on the ability of the tumor cells to express AGT independent of their expression levels before chemotherapy.
Detection of MGMT Promoter Hypermethylation
MS-PCR and MS-MLPA (using the average of the 3 MS-MLPA probes) were compared, and statistical evaluation identified a high agreement between both techniques in the detection of methylated MGMT sequences. In general, ratios below 0.25 correlate well with the detection of only unmethylated DNA by MS-PCR, whereas in cases showing ratios above 0.25 generally methylated DNA is detected (with or without unmethylated DNA). There is no clear cutoff MS-MLPA ratio to distinguish between identification of M and MU by MS-PCR in this series. As different CpGs are evaluated in both approaches, such differences might result from hypermethylation of only part of the CpGs. Furthermore, during MS-MLPA, methylation of individual CpGs is investigated, whereas multiple CpGs are located in the MS-PCR primers used (Figure 2). A negative effect of hypermethylation of only part of these CpGs on the MS-PCR efficiency is to be expected using the U as well as the M primer sets and might result in unsuccessful MS-PCR experiments or an underestimation of methylated CpGs.
After full optimization and changing to a newly developed bisulfite conversion kit, MS-PCR proved to be more efficient (successrate of 96%) than we initially expected the literature. Interestingly, a similar MS-PCR approach was recently described in a paper reporting a simplified laboratory validated assay for MGMT promoter hypermethylation testing,27 corroborating the value of this new bisulfite conversion kit. Nevertheless, MS-MLPA has still some significant advantages for the detection of DNA hypermethylation.
In general, p-DNA can be difficult to analyze at the molecular level owing to DNA fragmentation. As MLPA probes hybridize to a region of only 50–60 base pairs, p-DNA can be easily analyzed.19, 20 Furthermore, the (potentially) troublesome bisulfite conversion of unmethylated cytosines can be omitted in MS-MLPA using a methylation-sensitive digestion. Performing this digestion after hybridization of the probes prevents the use of a high amount of DNA or minute volumes during digestion. It results in complete digestion of all probes hybridized to unmethylated sequences, which are consequently not amplified during MS-MLPA PCR, in contrast to undigested probes hybridized to methylated sequences. The observation that digestion of the MS-specific probes is completely prevented when using (in vitro) methylated DNA shows that methylation of one of both strands (tumor DNA can be methylated whereas the complementary hybridized probe cannot) is sufficient. Additionally, as multiple probes (up to 45) can be included in a single MLPA experiment, the methylation status of different individual CpG dinucleotides can be evaluated simultaneously. By increasing the number of MGMT probes, the involvement of the specific CpGs can be further evaluated.
As shown in our titration experiments, MS-MLPA ratios are indicative of the degree of hypermethylation present in the sample. Quantitative analysis of MGMT has been described in gliomas using real-time MS-PCR.28 In contrast to MS-MLPA, these assays still require bisulfite conversion and specialized equipment that is not available in most molecular diagnostic laboratories. Furthermore, caution should be taken when choosing an internal control. For example, it is important to realize that the reported control gene β-actin is located in a chromosomal region (7p) that is frequently gained in high-grade gliomas (eg, GBMs), and it is therefore an inadequate control for glioma research. When working with tumor samples, one should keep in mind that all chromosomes can be affected by copy number changes, and that the use of multiple internal controls located on different chromosomes may improve the reliability of this approach. Fortunately, these problems do not occur when using MS-MLPA as the methylation ratio is calculated by comparing the total amount of DNA (M and U; undigested sample) with the amount of DNA present after the methylation-sensitive digestion, containing only the methylated fragments.
MGMT Promoter Hypermethylation in Gliomas
Twenty-nine patients with GBM were evaluated and the absence of, mild, moderate, and extensive MGMT hypermethylation were detected in 13 (45%), 10 (34%), 4 (14%), and 2 (7%) cases, respectively. Overall, in the majority of cases (79%), the absence of or only mild levels of MGMT promoter hypermethylation were detected, which might explain why TMZ shows only modest activity for GBMs. In oligodendroglial tumors (Os and OAs), moderate to extensive hypermethylation was detected in 4/8 cases. This is in line with a previous report showing a higher frequency of MGMT promoter hypermethylation in oligodendroglial tumors29 and correlates well with the observation that oligodendroglial tumors show a better response to TMZ than GBMs.30 Loss of 1p and 19q was detected previously in four of seven oligodendroglial tumors analyzed and interestingly, three of these were shown to contain moderate to extensive MGMT hypermethylation, suggesting a correlation between loss of 1p/19q and MGMT hypermethylation.
Methylation-mediated Silencing of MGMT and Sensitivity to Alkylating Agents
A first impression of the predictive value for response to chemotherapy of this MGMT MS-MLPA assay was obtained by evaluating a group of 20 GBM patients receiving TMZ treatment. Unfortunately, this group is still rather heterogeneous as it includes primary and secondary GBMs, patients who underwent gross total resection vs biopsy only, and also differences at the start of TMZ treatment (immediately following surgery vs at the time of progression). In this small study set, we found a trend toward longer survival with increasing MGMT hypermethylation ratio (average of all three probes). We should, however, keep in mind that, next to MGMT, other factors might also be involved, which is exemplified by case N460 showing an unexpectedly long survival (over 60 months) for a tumor that was classified as unmethylated (suggesting chemoresistance) by both MS-PCR and MS-MLPA. The actual predictive value of MGMT MS-MLPA needs to be further evaluated in large and more homogenous patient groups and is part of our ongoing investigations.
When performing clinical validation of the predictive value of this MGMT MS-MLPA assay, different aspects need to be addressed: (i) are specific CpG dinucleotides more relevant in predicting chemoresponse (ii) is there a linear correlation between increased ratios and increased survival or, can cut-off levels be identified that segregate responders from non responders (iii) what is the predictive value for the different chemotherapeutic (alkylating) agents and (iv) is MGMT hypermethylation indeed a poor prognostic factor for patients not receiving chemotherapy.
Genetic and Epigenetic Inactivation of MGMT
Genetic and epigenetic aberrations (eg, mutations or copy number changes vs hypermethylation) are intricately connected in cancer development. For example, it has been shown that aberrant promoter hypermethylation of one allele with mutation or loss of heterozygosity in the other allele, or methylation of both alleles, has the same effect, that is, functional inactivation of the gene.31, 32 Interestingly, MGMT hypermethylation is detected in part of the GBMs, whereas loss of chromosome 10 (including the MGMT gene at 10q26) is also a frequent event in these tumors.
Using MS-MLPA, we analyzed copy number changes and hypermethylation of the MGMT promoter. The distribution of MS-MLPA ratios was comparable for tumors with and without a loss of MGMT. As tumors with a loss showed MS-MLPA ratios along the entire range from 0 to 1, this suggests that tumors can be polyclonal in the methylation of their remaining allele. Although MS-MLPA ratios may be underestimated due to the presence of normal (U) ‘contaminating’ tissue in the tumor sample, data of the analyzed celllines (eg T98G) corroborate this observation. This is important from a clinical point of view, as it can be expected that, during chemotherapeutic treatment, cells with a hypermethylated MGMT promoter die, whereas the cells with an unmethylated promoter survive and ultimately lead to tumor progression and ‘acquired’ chemoresistance. So, whereas the detection of an unmethylated MGMT promoter next to methylated sequences is usually disregarded as originating from normal tissue, it may frequently reflect tumor heterogeneity. Comparing the degree of hypermethylation, as detected by MS-MLPA to the amount of normal cells present in the analyzed sample, provides an insight into the polyclonality of the tumors regarding MGMT hypermethylation. This latter information may be indicative of the duration of chemoresponse.
In Summary
Expression of AGT encoded by the MGMT gene results in chemoresistance when using alkylating agents, whereas hypermethylation of the MGMT promoter is associated with chemosensitivity, as it prevents expression. Because interpretation of immunohistochemical detection of AGT can be difficult, there is a need for evaluation of the MGMT methylation status. Our parallel analysis of MGMT promoter hypermethylation, using the newly developed MS-MLPA and the conventionally used MS-PCR, show high agreement. MS-MLPA analysis proved to be a reliable alternative for MS-PCR, which (1) gave reliable results in all samples, including p-DNAs, (2) provided semiquantitative data, (3) can be used to evaluate methylation status of multiple sequences simultaneously, and (4) allowed a combined copy number detection and methylation specific analysis. The potential value of MGMT hypermethylation evaluation using MS-MLPA was shown in a small group of patients with a GBM receiving TMZ chemotherapy. Further evaluation is, however, needed to establish its full clinical value in a large and more uniform group of patients. Especially the semiquantitative aspect of MS-MLPA may prove to be of great value in predicting (duration of) response to alkylating agents, as it is expected that the chemoresistant cells not showing MGMT hypermethylation will finally outgrow the chemosensitive cells harboring MGMT hypermethylation. Introduction of this MS-MLPA assay may not only be helpful for predicting response of gliomas to TMZ (as evaluated by us), but may also facilitate tailor-made treatment with other chemotherapeutic agents for a variety of tumors.
References
Drablos F, Feyzi E, Aas PA, et al. Alkylation damage in DNA and RNA—repair mechanisms and medical significance. DNA Repair (Amst) 2004;3:1389–1407.
Esteller M, Herman JG . Generating mutations but providing chemosensitivity: the role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene 2004;23:1–8.
Gerson SL . MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer 2004;4:296–307.
Srivenugopal KS, Yuan XH, Friedman HS, et al. Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-benzylguanine or 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochemistry 1996;35:1328–1334.
Delaney JC, Essigmann JM . Effect of sequence context on O(6)-methylguanine repair and replication in vivo. Biochemistry 2001;40:14968–14975.
Friedman HS, McLendon RE, Kerby T, et al. DNA mismatch repair and O6-alkylguanine-DNA alkyltransferase analysis and response to Temodal in newly diagnosed malignant glioma. J Clin Oncol 1998;16:3851–3857.
Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000;343:1350–1354.
Paz MF, Yaya-Tur R, Rojas-Marcos I, et al. CpG island hypermethylation of the DNA repair enzyme methyltransferase predicts response to temozolomide in primary gliomas. Clin Cancer Res 2004;10:4933–4938.
Nakagawachi T, Soejima H, Urano T, et al. Silencing effect of CpG island hypermethylation and histone modifications on O6-methylguanine-DNA methyltransferase (MGMT) gene expression in human cancer. Oncogene 2003;22:8835–8844.
Herman JG, Graff JR, Myohanen S, et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA 1996;93:9821–9826.
Warnecke PM, Stirzaker C, Song J, et al. Identification and resolution of artifacts in bisulfite sequencing. Methods 2002;27:101–107.
Dikow N, Nygren AO, Schouten JP, et al. Quantification of the methylation status of the PWS/AS imprinted region: comparison of two approaches based on bisulfite sequencing and methylation-sensitive MLPA. Mol Cell Probes 2007;21:208–215.
Nygren AO, Ameziane N, Duarte HM, et al. Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res 2005;33:e128.
Kleihues P, Cavenee WK . World health organization classification of tumours. Pathology and Genetics. Tumours of the Nervous System. International Agency for Research on Cancer (IARC) Press: Lyon, France, 2000.
Miller SA, Dykes DD, Polsky HF . A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acid Res 1988;16:1215.
Jeuken JWM, Sprenger SHE, Wesseling P, et al. Identification of subgroups of high-grade oligodendroglial tumors by comparative genomic hybridization. J Neuropathol Exp Neurol 1999;58:606–612.
Esteller M, Hamilton SR, Burger PC, et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 1999;59:793–797.
Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acid Res 2002;30:e57.
Jeuken JWM, Cornelissen S, Boots-Sprenger SHE, et al. Multiplex ligation-dependent probe amplification (MLPA): a diagnostic tool for simultaneous identification of different genetic markers in glial tumors. J Mol Diagnostics 2006;8:433–443.
van Dijk MC, Rombout PD, Boots-Sprenger SH, et al. Multiplex ligation-dependent probe amplification for the detection of chromosomal gains and losses in formalin-fixed tissue. Diagn Mol Pathol 2005;14:9–16.
Petrie A, Sabin C . Assessing agreement. In: Petrie A, Sabin C (eds). Medical Statistics at a Glance. Blackwell Science: Oxford, UK, 2004, pp 93–95.
Brell M, Tortosa A, Verger E, et al. Prognostic significance of O6-methylguanine-DNA methyltransferase determined by promoter hypermethylation and immunohistochemical expression in anaplastic gliomas. Clin Cancer Res 2005;11:5167–5174.
Nakasu S, Fukami T, Baba K, et al. Immunohistochemical study for O6-methylguanine-DNA methyltransferase in the non-neoplastic and neoplastic components of gliomas. J Neurooncol 2004;70:333–340.
Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005;352:997–1003.
Alvino E, Castiglia D, Caporali S, et al. A single cycle of treatment with temozolomide, alone or combined with O(6)-benzylguanine, induces strong chemoresistance in melanoma cell clones in vitro: role of O(6)-methylguanine-DNA methyltransferase and the mismatch repair system. Int J Oncol 2006;29:785–797.
Margison GP, Povey AC, Kaina B, et al. Variability and regulation of O6-alkylguanine-DNA alkyltransferase. Carcinogenesis 2003;24:625–635.
Cankovic M, Mikkelsen T, Rosenblum ML, et al. A simplified laboratory validated assay for MGMT promoter hypermethylation analysis of glioma specimens from formalin-fixed paraffin-embedded tissue. Lab Invest 2007;87:293–297.
Maxwell JA, Johnson SP, Quinn JA, et al. Quantitative analysis of O6-alkylguanine-DNA alkyltransferase in malignant glioma. Mol Cancer Ther 2006;5:2531–2539.
Mollemann M, Wolter M, Felsberg J, et al. Frequent promoter hypermethylation and low expression of the MGMT gene in oligodendroglial tumors. Int J Cancer 2005;113:379–385.
van den Bent MJ, Taphoorn MJ, Brandes AA, et al. Phase II study of first-line chemotherapy with temozolomide in recurrent oligodendroglial tumors: the European Organization for Research and Treatment of Cancer Brain Tumor Group Study 26971. J Clin Oncol 2003;21:2525–2528.
Jones PA, Laird PW . Cancer epigenetics comes of age. Nat Genet 1999;21:163–167.
Herman JG, Baylin SB . Gene silencing in cancer in association with promoter hypermethylation. N Eng J Med 2003;349:2042–2054.
Acknowledgements
This project was sponsored by the Dutch Cancer Society (Koningin Wilhelmina Fonds: KUN 2004-3143) and the Pauline van Everdingen Foundation. We thank Jan Schouten from MRC-Holland for developing and kindly supplying the MGMT MS-MLPA kits and for helpful discussions. We also thank Dr Hans Bernsen (CWZ), Dr Mathé Prick (CWZ) and Dr Anja Gijtenbeek (RUNMC) for providing the clinical information, and the neurosurgeons of the RUNMC and CWZ for their continuous collaboration.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jeuken, J., Cornelissen, S., Vriezen, M. et al. MS-MLPA: an attractive alternative laboratory assay for robust, reliable, and semiquantitative detection of MGMT promoter hypermethylation in gliomas. Lab Invest 87, 1055–1065 (2007). https://doi.org/10.1038/labinvest.3700664
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700664
Keywords
This article is cited by
-
A novel analytical model of MGMT methylation pyrosequencing offers improved predictive performance in patients with gliomas
Modern Pathology (2019)
-
ColPortal, an integrative multiomic platform for analysing epigenetic interactions in colorectal cancer
Scientific Data (2019)
-
Methylated DNA/RNA in Body Fluids as Biomarkers for Lung Cancer
Biological Procedures Online (2017)
-
Correlation of c-MET Expression with PD-L1 Expression in Metastatic Clear Cell Renal Cell Carcinoma Treated by Sunitinib First-Line Therapy
Targeted Oncology (2017)
-
Primary and recurrent diffuse astrocytomas: genomic profile comparison reveals acquisition of biologically relevant aberrations
Molecular Cytogenetics (2016)
