
Abstract
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of gastrointestinal tract. GISTs range from benign indolent neoplasms to highly malignant sarcomas. Gain-of-function mutations of tyrosine kinase receptors, KIT or PDGFRA, have been identified in most GISTs. In this study, we report 36 GIST patients whose tumors had homozygous KIT exon 11 mutations detected by direct sequencing of PCR products. Loss of heterozygosity in KIT locus and other chromosome 4 loci were documented in majority of these tumors. However, fluorescence in situ hybridization with KIT locus-specific probe and chromosome 4 centromeric enumeration probe showed no evidence of KIT hemizygosity in a majority of analyzed cases. These findings are consistent with duplication of chromosome 4 with KIT mutant allele. Homozygous KIT exon 11 mutations were found in 33 primary tumors and 7 metastatic lesions. In two cases, shift from heterozygosity to homozygosity was documented during tumor progression being present in metastases, but not in primary tumors. Among primary GISTs, there were 16 gastric, 18 intestinal and 2 from unknown locations. An average primary tumor size was 12 cm and average mitotic activity 32/50 HPFs. Out of 32 tumors 29 (90.6%) with complete clinicopathologic data were diagnosed as sarcomas with more than 50% risk of metastatic disease, and 26 of 29 patients with follow-up had metastases or died of disease. An average survival time among pre-imatinib patients, who died of the disease was 33.4 months. Based on these findings, we conclude that presence of homozygous KIT exon 11 mutations is associated with malignant course of disease and should be considered an adverse prognostic marker in GISTs.
Similar content being viewed by others


Main
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of gastrointestinal (GI) tract. GISTs can occur in any part of GI tract, but are most frequently found in the stomach and small intestine. GISTs show spindle or epithelioid cell morphology, and occasionally pleomorphic features. A great majority of GISTs express KIT.1 KIT expression links GISTs to interstitial cells of Cajal, hypothetic GIST progenitor cells.2, 3
KIT or PDGFRA gain-of-function mutations have been identified in a great majority of GISTs and are considered to be one of the first molecular events in their pathogenesis; these mutations lead to pathological activation of KIT or PDGFRA signaling pathways.3, 4 Both KIT and PDGFRA belong to the type III tyrosine kinase receptor family and play an important role in different cell functions including cell proliferation.5
Tumor size and mitotic activity are the most important prognostic parameters in GISTs.1 However, recent studies have shown that molecular genetic markers, such as the type of KIT mutation, might have prognostic value. One study reported gastric tumors with KIT exon 11 deletions to have more malignant clinical outcome than the ones with point mutations.6 Other studies have shown that GISTs with KIT 1690_1695del (Tyr557_Lys558del at the protein level) have a significantly worse prognosis than ones with other KIT exon 11 mutations.7, 8
Since 2000, the tyrosine kinase inhibitor, imatinib mesylate (Gleevec®, Novartis, USA) has been successfully used in the treatment of clinically advanced and metastatic GISTs.9 The type of KIT or PDGFRA mutation indicates tumor responsiveness to imatinib treatment. Whereas KIT exon 11 (juxtamembrane domain) mutant GISTs achieve the highest level of response, tumors with Asp842Val, the most common PDGFRA exon 18 (tyrosine kinase domain) mutation, do not respond to imatinib mesylate treatment.10 A more recent study showed that GISTs with KIT exon 9 (extracellular domain) mutations required two times higher dose of imatinib mesylate to achieve a response similar to that observed in KIT exon 11 mutant tumors.11
Although GISTs with homozygous KIT exon 11 mutations have been sporadically reported in the literature, the clinicopathologic profile of such tumors is not known.12, 13, 14, 15, 16, 17, 18, 19 In this study, we report a series of 36 primary and metastatic GISTs with homozygous KIT exon 11 mutations and review the clinicopathologic profile and natural history of such tumors before the availability of tyrosine kinase inhibitors. Moreover, we report responsiveness of GISTs with homozygous KIT exon 11 mutations to imatinib treatment.
Materials and methods
Material
Formalin-fixed, paraffin embedded (FFPE) samples of tumor and corresponding normal tissue were retrieved from the files of the following institutions: Armed Forces Institute of Pathology (AFIP), Washington DC, USA; M. Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Warszawa, Poland; Haartman Institute of the University of Helsinki, Helsinki, Finland; University Hospital Northern Norway, Tromso, Norway; Otto-von-Guericke University, Magdeburg, Germany; Collegium Medicum of the Jagiellonian University, Krakow Poland; Department of Pathology, Pomeranian Medical University, Szczecin, Poland; and University College Hospital, Ibadan, Nigeria. Demographic, clinical and follow-up data were obtained according to the Institutional Review Board approval.
Tumors were diagnosed as GISTs using previously established histological and immunohistochemical criteria.1 Based on tumor size and mitotic activity, primary GISTs were classified into eight prognostic groups, indicating likelihood of malignant behavior (Table 1). Response to imatinib treatment was evaluated following the criteria provided by RECIST (response evaluation criteria in solid tumors) guidelines.21
KIT Mutation Status
GIST KIT mutation databases at the Department of Soft Tissue Pathology, AFIP and at the Department of Molecular Biology, M. Sklodowska-Curie Memorial Cancer Center were screened for tumors with homozygous KIT exon 11 mutations. These mutations were previously identified at the DNA level by PCR amplification and direct sequencing, following published procedures.22 In two cases, ‘hot spots’ in KIT exon 13, 14 and 17 were evaluated for secondary mutations acquired during imatinib mesylate-based treatment. The following primers were used for PCR amplification: CK13.5F (76271_76290) and CK13.2.1R (76369_76388) for exon 13; CK14.7F (77544_77563) and 14.6R (77670_77690) for exon 14; CK17.1F (81351_81370) and CK17.2R (81451_81570) for exon 17. PCR conditions were standard with annealing temperatures 55oC in all reactions.
Nomenclature of the mutations was based on the recommendations of Human Genome Mutation Society (www.hgvs.org). Mutations at the protein level were deduced with the assumption that all changes identified at the genomic level involved one allele. The following KIT (HSU63834, XO6182) and PDGFRA (ACO98587) reference sequences were obtained from National Center for Biotechnology Information (NCBI) at www.ncbi.nlm.nih.gov.
Loss of Heterozygosity Studies
Loss of heterozygosity (LOH) was evaluated by PCR amplification of three chromosome 4q (D4S3045, D4S1619, D4S392) and one chromosome 4p (D4S2950) microsatellite markers. Marker positions and primer sequences were obtained from human genome microsatellite marker databases linked to the NCBI webpage at www.ncbi.nlm.nih.gov. PCR amplifications were performed using the standard conditions recommended by Applied Biosystems (www.appliedbiosystems.com). PCR products were analyzed on ABI PRISM® 310 Genetic Analyzer, following the Applied Biosystems procedure. A ratio of the peak high values (fluorescence intensity) between the longer and shorter allele was calculated for normal and tumor tissues. To obtain the LOH value, an allele ratio from normal tissue was divided by an allele ratio from tumor tissue. The values ≤0.5 and ≥1.5 were considered to indicate LOH, as recommended by PE Biosystems and reported previously.23 The borderline values >0.5 <0.6 and >1.5<1.6 were considered to represent a partial LOH.
Twelve KIT and two PDGFRA single-nucleotide polymorphisms (SNPs) were evaluated by PCR amplification and direct sequencing. Locations of SNPs were obtained from databases available at NCBI. In addition, during the screening, two previously unreported SNPs in exon 10 and exon 17 were identified and evaluated as well. The frequencies of these two SNPs were 10 and 6.7%, respectively, and were based on 30 normal DNA samples from unrelated healthy individuals tested in this study. Primer positions and PCR conditions used to amplify KIT and PDGFRA SNPs are listed in Table 2. All KIT and PDGFRA SNPs evaluated in this study are listed in Table 3.
Also, six SNPs reported in Huntington's Disease gene (HD), located at 4p16.3, were evaluated in 18 cases. Primer sequences and PCR conditions used for HD SNPs studies are listed in Table 4.
All microsatellite marker- and SNP-based LOH studies were carried out independently in different laboratories.
FISH Studies
Interphase FISH studies were performed on standard 5-μm sections of FFPE tissues prepared for hybridization using SPoT-Light Cell Pretreatment Kit, following manufacturer's protocol (Zymed, CA, USA). BAC clone RP11-959G16 containing the full length of KIT was used as the locus-specific probe (LSP), together with the chromosome 4 centromeric enumeration probe (CEP) (Vysis Inc., Downers Grove, IL, USA). The probes were assessed individually or simultaneously following previously reported procedures.11 The images were analyzed with Zeiss Axioplan 2 (Zeiss, Germany) fluorescence microscope and captured by cooled black-and-white charged-couple device camera coupled with Isis FISH Imaging System version 5.1 software (Metasystems, Germany).
In each case, at least 100 intact, nonoverlapping nuclei were chosen for scoring fluorescent signals. The percentage of tumors nuclei containing 0, 1, 2, 3, 4, >4 signals were calculated for each probe. Based on a previously published study,24 the following criteria for FISH anomalies were applied: (1) abnormal gain required 10% nuclei with three or more signals; (2) abnormal loss required ≥65% nuclei with 0 or 1 signal. All FISH analyses were carried out on coded slides without knowledge of other data.
Statistical Studies
Prognostic comparative data were analyzed using Kruskal-Wallis test. All statistical tests were two-sided and 5% level of significance was used.
Results
KIT Mutation Studies
Screening of 700 KIT exon 11 mutant GISTs from the AFIP database and 32 KIT exon 11 mutant GISTs from the M. Sklodowska-Curie Memorial Cancer Center database revealed 27 (3.86%) and 9 (28.1%) homozygous mutations, respectively. There were 27 (75%) deletions (del) or deletion-insertions (delins), 7 (19.4%) single-nucleotide substitutions (point mutations (pm)) and 2 (5.6%) duplications (dup). Deletions ranged from 3 to 57 nucleotides and mostly involved 5′ part of KIT exon 11. The most common deletion, 1690_1695del leading at the protein level to Tyr557_Lys558del, was found in three cases. Deletions 1756_1758del (Asp579del) and 1682_1738del (Glu554_Pro573delinsAla) were identified in two cases each. Remaining deletions and deletion-insertions were unique. Substitutions were found at codons 557 (n=2), 560 (n=4) and 576 (n=1). Two duplications consist of in-frame repeat of 6 and 39 nucleotides in the 3′ part of KIT exon 11. Genomic sequences of all homozygous mutations and deduced mutant KIT protein sequences are listed in Table 4. Representative examples of direct sequencing of KIT exon 11 PCR products are shown in Figure 1a.

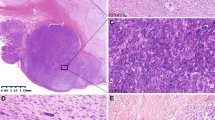
(a) Example of direct sequencing of KIT PCR amplification products in Case 35. Upper panel primary tumor, lower panel metastatic lesion; double and single arrows indicate heterozygous and homozygous point mutations, respectively. (b) Example of LOH detected by evaluation of microsatellite D4S1619 marker in Case 35; arrow indicates lost allele. (c) Example of FISH with KIT LSP (red signal) and CEP 4 (green signal) in Case 17. Note increased number of KIT and centromeric signals, indicating increased copy number of chromosome 4. (d, e) Representative histological and immunohistochemical images of GISTs analyzed in this study. (d) Malignant intestinal GIST with spindle cell morphology (Case 22). (e) Strong KIT immunoreactivity in a malignant gastric GISTs with spindle cell morphology (Case 1).
Two cases with metastatic lesions resistant to imatinib treatment were screened for secondary KIT mutations in exon 13, 14 and 17. A 1982T>C substitution leading to heterozygous Val654Ala mutation was identified in one case.
LOH Studies
Normal tissue was available in 29 of 36 cases. These cases were evaluated, for LOH at chromosome 4q loci, using 3 microsatelite markers and 14 KIT, PDGFRA SNPs and for LOH at chromosome 4p loci, using 1 microsatellite marker and 6 HD SNPs. Successful amplification was obtained in 531 (89.9%) of 591 analyses. LOH was identified in 174 (90.2%) of 193 informative microsatelite and SNP loci. A representative microsatellite-based LOH assay is shown in Figure 1b. Results of all LOH studies are summarized in Figure 2.

Summary of LOH and FISH studies. Single assay is represented by a circle. Gray and black colors indicate constitutional heterozygosity with retention of both allele and loss of one allele, respectively. White color indicates homozygosity (non-informative loci). Gray squares with black circles indicate borderline LOH values. Abbreviations: P: primary tumor; M: metastatic tumor; nd: not done; na: no PCR amplification products. Tumors with ≥65% of nuclei with none or one FISH signal are indicated by rectangles.
In 28 tumors, multiple informative SNPs and microsatellite markers were lost indicating possible loss of the entire copy of chromosome 4. In two tumors (Cases 3 and 30), one to four KIT SNPs clustered between introns 1 and 8 remained polymorphic. Also, the two PDGFRA SNPs clustered in intron 17 and exon 18 remained polymorphic in one case (Case 18). Moreover, in one tumor (Case 11), lack of LOH was documented at D4S2950, chromosome 4p microsatellite marker and in another tumor (Case 30), two HD SNPs were preserved.
In one tumor (Case 2), LOH was detected only in KIT intron 4 and 8 and at chromosome 4p loci. In this case, microsatellite markers telomeric to KIT/PDGFRA locus remained polymorphic, indicating retention of a large portion of chromosome 4q.
FISH Studies
FISH with KIT LSP was successful in 17 tumors. A representative FISH image is shown in Figure 1c. In three (17.7%) tumors, no or one signal was found in ≥65% of nuclei, suggesting loss of KIT locus. In 14 (82.3%) tumors, the percentage of nuclei with ≥2 KIT LSP signals ranged from 45 to 99%, suggesting either a diploid karyotype or abnormal gain. Moreover, in five of these tumors, a ratio of KIT LSP to CEP4 FISH signals was 1 or close to 1. In Case 3, FISH analysis of the primary and subsequent metastatic lesion revealed a decrease of the percentage of nuclei with 1 signal from 65% in primary tumors to 24% in metastatic one, and an increase of the percentage of nuclei with >2 signals from 5% in primary tumor to 34% in metastatic tumors.
Demographic, Clinical and Pathologic Features
All demographic, clinical and pathologic data are summarized in Table 5. The patient age varied from 37 to 80 years with median age of 62 years. The male to female ratio was 17:19. There were 16 gastric, 15 small intestinal, 2 colonic and 1 rectal GIST. The primary location could not be established for two tumors.
Thirty (83.3%) tumors had spindle cell morphology. There were two and three epithelioid gastric and intestinal tumors, respectively. One gastric GIST revealed mixed histology with both spindle and epithelioid components. KIT (CD117) expression was documented immunohistochemically in all analyzed cases. Representative histological and immunohistochemical images are shown in Figure 1d and e.
The size of primary tumors ranged from 2 to 30 cm (median 12.3 cm). Twenty-nine of 34 (85.3%) primary GISTs with known size of the primary tumor were >5 cm in diameter. Mitotic activity in the primary tumors varied from 1 to >100 per 50/HPF with an average of 32 mitoses per 50 HPF.
Complete or partial clinical and follow-up data were available in 33 cases (Table 6). Twenty-eight (84.5%) patients developed metastasis or died of disease. The average survival time for the 14 pre-imatinib patients who died of disease was 33.4 months. Based on previously published criteria (Table 1), three GISTs without follow-up data represented malignant tumors with 55 to 85% chance of developing metastatic diseases. Also, two patients that died of unknown causes had malignant tumors with higher than 55% risk of metastasis. Only 3 patients diagnosed with GIST with low to moderate risk of metastases were alive with follow-up ranging from 7 to 15 months (average 11.6 months).
Eight patients with advanced, disseminated GISTs were treated with imatinib mesylate. Initially, a partial response and stable disease were seen in six and one cases, respectively. There was insufficient clinical follow-up in one case. Subsequently, a patient with stable disease and three patients with partial response developed progressive disease (Table 7).
The metastatic tumors resistant to imatinib mesylate treatment were available for molecular studies (Case 9 and 13). Both lesions were screened for secondary mutations affecting KIT exons 13, 14 and 17. A heterozygous 1982T>C substitution in KIT exon 13, leading to Val654Ala mutation at the protein level was identified in Case 13.
The average survival time for the three patients treated with imatinib mesylate, who died of disease was 27 months. Three other patients remained in partial response at the time of this study.
Statistical Studies
Risk of progressive disease in gastric and intestinal homozygous KIT exon 11 mutant GISTs was compared with risk of progressive disease in the cohorts of gastric and intestinal tumors with heterozygous KIT exon 11 mutations or without determined mutational status. Previously published data on gastric and small intestinal GISTs were included in statistical studies.6, 20 All results are summarized in Tables 8 and 9.
The risk of progressive disease was significantly higher in GISTs with homozygous KIT exon 11 deletion/deletion-insertions than in GISTs with heterozygous KIT exon 11 deletions/deletion-insertions or without determined mutational status. No difference in risk of progressive disease was detected when small intestinal GISTs with homozygous single nucleotide substitutions (point mutations) were compared with heterozygous mutants. However, the cohort of tumors with homozygous single-nucleotide substitutions was relative small and included only four cases.
Discussion
GISTs encompass a spectrum of mesenchymal tumors from benign, indolent lesions to highly malignant sarcomas.1 GISTs are believed to originate from interstitial cells of Cajal or their precursor cells being driven by gain-of-function KIT and PDGFRA mutations.2, 3, 4
Most KIT mutations are heterozygous. However, in some cases, only the mutant allele can be identified by direct sequencing of PCR amplification products. There are several possible explanations for such findings. These include the presence of the same mutation in both alleles (truly homozygous mutation), presence of KIT-mutant (MT) and absence of KIT-(wild type) WT allele (hemizygous mutation), and selective amplification of mutant KIT locus or polysomy of KIT-MT chromosome 4.
In this study, we have examined the nature of homozygous KIT exon 11 mutations, as detected by direct sequencing of PCR products. Lack of polymorphism at multiple polymorphic sites indicated loss of KIT-WT chromosome 4. However, dual-color FISH using KIT LSP and chromosome 4 CEP showed no evidence of KIT deletion or loss of one copy of chromosome 4 in majority of analyzed tumors. Also, shift from chromosome 4 monosomy in primary tumor to disomy in metastatic lesion was documented by FISH in one case. Together, these data suggested that a loss of KIT-WT chromosome 4 is followed by a duplication of KIT-MT chromosome 4. A similar molecular mechanism was previously reported in two KIT exon 13 mutant GISTs.25
In one tumor (Case 2), multiple polymorphic markers retained polymorphism and only those located in the vicinity of KIT exon 11 showed homozygosity. The presence of genetic changes other than loss of entire chromosome 4 should be considered in this case. For example, coexistence of a large KIT deletion in another KIT allele or complex genomic rearrangements could explain this finding. Also, in a few cases, selected SNPs and microsatellite markers remained polymorphic, indicating retention of genetic material from KIT-WT chromosome 4, most likely involved in complex genetic rearrangements. Identification of such changes was beyond the scope of our investigation based on archival FFPE material.
Numeric changes of chromosome 4 copies identified by classical karyotyping or FISH are relatively rare among GISTs.24, 26 Also, comparative genomic hybridization studies have failed to identify losses or gains of genetic material from chromosome 4 in GISTs.27 The current study showed that loss of KIT-WT chromosome 4 could be masked by duplication of KIT-MT chromosome 4, a molecular event that can not be identified by classical karyotyping or comparative genomic hybridization.
In the AFIP KIT mutation database, homozygous KIT exon 11 mutations represented only a small fraction, approximately 4%, of all KIT exon 11 mutations. Also, a recent study based on population of Northern Norway reported homozygous KIT mutations in 4.5% (4 of 89) analyzed GISTs.28 However, one study based on 322 GISTs, including 127 malignant tumors from a clinical imatinib mesylate trial, identified homozygous KIT mutations in 17.8% of analyzed cases.10 Also, a substantially higher frequency (28%) was seen among cases contributed to our study by Sklodowska-Curie Memorial Cancer Center, which specializes in the treatment of advanced GISTs. The higher frequency of homozygous KIT exon 11 mutations in the materials from clinical trials and large cancer centers clearly reflects selection bias and further support the idea that homozygous mutations are enriched among patients with malignant, highly advanced tumors.
A combination of clinicopathologic features such as tumor size and mitotic activity is considered to be the most important prognostic parameter in GISTs.1 However, differences in clinical outcome between GISTs from different locations have been reported.1 In general, small intestinal tumors tend to follow a more malignant course of disease than gastric ones.20 In this series, almost entirely based on malignant GISTs, small intestinal tumors were overrepresented, if compared with the data reported by population-based studies.29, 30 This confirms previous observations that tumors from small intestinal locations are enriched in cohorts of malignant GISTs.1
Recent studies have also shown that the type of KIT mutation might correlate with the clinical outcome. Gastric GISTs with KIT exon 11 deletions have more malignant clinical outcomes than the ones with point mutations.6 In the present study, none of the 16 gastric GISTs had KIT exon 11 point mutations. In contrast, point mutations were found in 31.6% of intestinal GISTs. Previous studies have shown that KIT codons 557_558 deletion indicates unfavorable prognosis in GISTs.7, 8 This deletion was identified in two malignant gastric and one small intestinal/mesenteric GIST, in this study. Duplications in the 3′ end of KIT exon 11 have been linked to gastric GISTs with rather benign clinical outcome.31, 32 Contrary to previous observations, two malignant tumors with such mutations including one of intestinal origin were reported in this study.
The current series of 36 GISTs showed a strong association between the presence of homozygous KIT exon 11 mutations and malignant clinical outcome. Tumors with such mutations generally had histologic features of sarcomas and developed intra-abdominal and liver metastases in the majority of cases. Therefore, the presence of homozygous KIT mutations was coupled with overall malignant features. However, GISTs with homozygous deletion/deletion-insertions showed a significantly higher risk of developing metastases than heterozygous mutants from similar locations.
In most cases, homozygous KIT mutations were already found in primary tumors. However, a great majority of these GISTs were at an advanced stage of disease, often with metastases. In two cases, a shift from heterozygosity to homozygosity was seen in metastatic lesions. This might indicate that KIT-MT(+)/KIT-WT(−) clones have a higher metastatic potential than KIT-MT(+)/KIT-WT(+) clones and supports the hypothesis that the presence of KIT-WT allele can moderate the effect of a KIT-MT allele. Also, two separate studies have reported two and three GISTs with a shift from heterozygosity to homozygosity seen only in metastatic lesions.11, 19 Thus, a shift from heterozygosity to homozygosity might be acquired during disease progression and accumulation of secondary genetic changes. However, one study reported homozygous KIT exon 11 mutations in 2 (15.4%) of 13 incidental, <1 cm GISTs.14 These findings could suggest that a shift from heterozygosity to homozygosity can also occur at an early stage of tumor development. However, two recent, separate studies failed to find homozygous KIT exon 11 mutations among 16 minimal GISTs (Agaimy personal communication).29, 33 In our series, no minimal GISTs with homozygous KIT exon 11 mutations were identified; however, two relatively smaller tumors (Cases 14 and 27) with low risk of developing progressive disease were reported. It is possible that homozygosity detected in small, benign GISTs differs in nature from that found in malignant tumors. Further studies employing different molecular techniques and based on a large number of cases are necessary to clarify this issue.
Familial GIST syndrome is associated with germline KIT mutation and development of multiple GISTs. Two studies have reported homozygous KIT mutations in large, malignant tumors diagnosed in familial GIST patients, whereas smaller, benign lesions have remained heterozygous.30, 34 Recently, we have identified a homozygous 1756_1758del leading to the loss of KIT codon 579 in a tumor from a patient with familial GIST syndrome. However, this tumor behaved indolently over 16 years.35 These findings suggest that the behavior of familial GISTs with homozygous KIT mutations may vary.
An amplification of KIT and PDGFRA has been reported in gliomas and shown to be more frequent in anaplastic and recurrent tumors than in low-grade lesions.36, 37 Also, selective KIT amplification leading to KIT overexpression has been reported in the seminoma subtype of testicular germ cell tumors and is linked to the progression of carcinoma in situ lesion to seminoma.38 In GISTs, amplification of the KIT/PDGFRA locus or selective amplification of KIT appears to be an extremely rare molecular event and has been reported only in a few cases.11, 39 Also, in our series, the KIT locus was not amplified. However, in the majority of analyzed GISTs, at least two copies of mutated KIT were present in a substantial number of tumor cells due to the duplication of KIT-MT chromosome 4.
In one study, a shift from heterozygosity to homozygosity was observed in two tumors at the time of resistance to imatinib; however, the resistance was also associated with secondary KIT mutation and it was unclear whether homozygosity or secondary mutation contributed to insensitivity to imatinib mesylate in these cases.19 In another study, KIT LOH was detected in highly cellular areas in the primary lesion and in liver metastasis resistant to imatinib.11
In our series, four of seven cases treated with imatinib mesylate developed progressive disease after relatively short time of 16–18 months. Moreover, the average survival time for the patients treated with imatinib mesylate, who died of the disease, was not different from the one calculated for the pre-imatinib patients. A second heterozygous KIT mutation, 1982T>C (Val654Ala) identified in one case, indicated that mutation related to resistance to imatinib mesylate treatment arose after chromosome 4 duplication. Although, secondary KIT mutation could account for tumor resistance, loss of KIT-WT allele, followed by duplications of KIT-MT allele and possibly increased of KIT-MT expression might also contribute to a more malignant clinical behavior and lower sensitivity to imatinib mesylate treatment. However, further clinicopathologic studies based on larger number of cases are necessary to verify this hypothesis.
In summary, this study documents the loss of KIT-WT allele and duplication of KIT-MT allele as molecular mechanisms leading to a shift from heterozygosity to homozygosity in a subset of GISTs. The risk of progressive disease was significantly higher among gastric and small intestinal GISTs with homozygous KIT exon 11 deletion/deletion-insertions than in tumors with heterozygous KIT exon 11 mutations. Also, follow-up data showed that the presence of such KIT exon 11 mutation represents a sign of disease progression and is associated with malignant course of disease. Thus, detection of homozygous KIT exon 11 mutations should be considered an additional adverse prognostic marker in GISTs.
References
Miettinen M, Lasota J . Pathology and prognosis of gastrointestinal stromal tumors at different sites. Semin in Diagn Pathol 2006;23:70–83.
Kindblom LG, Remotti HE, Aldenborg F, et al. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 1998;153:1259–1269.
Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998; 279:577–580.
Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708–710.
Pawson T . Regulation and targets of receptor tyrosine kinases. Eur J Cancer 2002;38 (Suppl 5):S3–S10.
Miettinen M, Sobin LH, Lasota J . Gastrointestinal stromal tumors (GISTs) of the stomach – a clinicopathologic, immunohistochemical and molecular genetic study of 1756 cases with long-term follow-up. Am J Surg Pathol 2005;29:52–68.
Wardelmann E, Losen I, Hans V, et al. Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. Int J Cancer 2003;106:887–895.
Martin J, Poveda J, Llombart-Bosch A, et al. Deletions affecting codons 557–558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: a study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol 2005;23:6190–6198.
Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472–480.
Corless CL, Fletcher JA, Heinrich MC . Biology of gastrointestinal stromal tumors. J Clin Oncol 2004;22:3813–3825.
Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumors. Eur J Cancer 2006;42:1093–1103.
Ernst SI, Hubbs AE, Przygodzki RM, et al. KIT mutation portends poor prognosis in gastrointestinal stromal/smooth muscle tumors. Lab Invest 1998;78:1633–1636.
Li QS, O’Leary TJ, Sobin LH, et al. Analysis of KIT mutation and protein expression in fine needle aspirates of gastrointestinal stromal/smooth muscle tumors. Acta Cytol 2000;44:981–986.
Corless CL, McGreevey L, Haley A, et al. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol 2002;160:1567–1572.
Emile JF, Theou N, Tabone S, et al. Clinicopathologic, phenotypic, and genotypic characteristics of gastrointestinal mesenchymal tumors. Clin Gastroenterol Hepatol 2004;2:597–605.
Hou YY, Tan YS, Sun MH, et al. C-kit gene mutation in gastrointestinal stromal tumors. World J Gastroenterol 2004;10:1310–1314.
Debiec-Rychter M, Cools J, Dumez H, et al. Mechanisms of resistence to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroentereology 2005;128:270–279.
Cho S, Kitadai Y, Yoshida S, et al. Deletion of the KIT gene is associated with liver metastasis and poor prognosis in patients with gastrointestinal stromal tumor in the stomach. Int J Cancer 2006;28:1361–1367.
Kikuchi H, Yamashita K, Kawabata T, et al. Immunohistochemical and genetic features of gastric and metastatic liver gastrointestinal stromal tumors: sequential analyses. Cancer Sci 2006;97:127–132.
Miettinen M, Makhlouf H, Sobin LH, et al. Gastrointestinal stromal tumors (GISTs) of the jejunum and ileum: a clinicopathologic, immunohistochemical and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol 2006;30:477–489.
Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 2000;90:205–216.
Lasota J, Wozniak A, Sarlomo-Rikala M, et al. Mutations in exons 9 and 13 of KIT gene are rare events in gastrointestinal stromal tumors. A study of two hundred cases. Am J Pathol 2000;157:1091–1095.
Canzian F, Salovaara R, Hemminki A, et al. Semiautomated assessment of loss of heterozygosity and replication error in tumors. Cancer Res 1996;56:3331–3337.
Debiec-Rychter M, Lasota J, Sarlomo-Rikala M, et al. Chromosomal aberrations in malignant gastrointestinal stromal tumors: correlation with c-KIT gene mutation. Cancer Genet Cytogenet 2001;128:24–30.
Lux ML, Rubin BP, Biase TL, et al. KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 2000;156:791–795.
Gunawan B, Bergmann F, Hoer J, et al. Biological and clinical significance of cytogenetic abnormalities in low-risk and high-risk gastrointestinal stromal tumors. Human Pathol 2002;33:316–321.
El Rifie W, Sarlomo-Rikala M, Andersson LC, et al. DNA sequence copy number changes in gastrointestinal stromal tumors: tumor progression and prognostic significance. Cancer Res 2000;60:3899–3903.
Steigen SE, Eide TJ, Wasag B, et al. Mutations in gastrointestinal stromal tumors—a population based study from northern Norway. APMIS 2007;115:69–72.
Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol 2007;31:113–120.
O’Riain C, Corless CL, Heinrich MC, et al. Gastrointestinal stromal tumors. Insights from a new familial GIST kindred with unusual genetic and pathologic features. Am J Surg Pathol 2005;29:1680–1683.
Antonescu CR, Sommer G, Sarran L, et al. Association of KIT exon 9 mutation with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clin Cancer Res 2003;9:3329–3337.
Lasota J, Dansonka-Mieszkowska A, Stachura T, et al. Gastrointestinal stromal tumors with internal tandem duplications in 3’ end of KIT juxtamembrane domain occur predominantly in stomach and generally seem to have a favorable course. Mod Pathol 2003;16:1257–1264.
Daum O, Grossmann P, Vanecek T, et al. Diagnostic morphological features of PDGFRA-mutated gastrointestinal stromal tumors: molecular genetic and histologic analysis of 60 cases of gastric gastrointestinal stromal tumors. Annals of Diagnostic Pathology 2007;11:27–33.
Kim HJ, Lim SJ, Park K, et al. Multiple gastrointestinal stromal tumors with a germline c-kit mutation. Pathol Int 2005;55:655–659.
Lasota J, Miettinen M . A new familial GIST identified. Am J Surg Pathol 2006;30:1342.
Joensuu H, Puputti M, Sihto H, et al. Amplification of genes encoding KIT PDGFRα and VEGFR2 receptor tyrosine kinases is frequent in glioblastoma multiforme. J Pathol 2005;207:224–231.
Puputti M, Tynninen O, Sihto H, et al. Amplification of KIT PDGFRA VEGFR2 and EGFR in Gliomas. Mol Cancer Res 2006;4:927–934.
McIntyre A, Summersgill B, Grygalewicz B, et al. Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res 2005;65:8085–8089.
Tabone S, Theou N, Wozniak A, et al. KIT overexpression and amplification in gastrointestinal stromal tumors (GISTs). Biochim Biophys Acta 2005;1741:165–172.
Acknowledgements
The opinions and assertions contained herein are the expressed views of the authors and are not to be construed as official or reflecting the views of the Departments of the Army or Defense. This study was partially supported by the American Registry of Pathology. B.W. was supported by the grant from the Foundation for Polish Science.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lasota, J., vel Dobosz, A., Wasag, B. et al. Presence of homozygous KIT exon 11 mutations is strongly associated with malignant clinical behavior in gastrointestinal stromal tumors. Lab Invest 87, 1029–1041 (2007). https://doi.org/10.1038/labinvest.3700628
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700628
Keywords
This article is cited by
-
Clinicopathological and mutational characteristics of primary double mutant gastrointestinal stromal tumor: a single center study with review of the literature
BMC Cancer (2023)
-
The secondary KIT mutation p.Ala510Val in a cutaneous mast cell tumour carrying the activating mutation p.Asn508Ile confers resistance to masitinib in dogs
BMC Veterinary Research (2020)
-
KIT mutation analysis in mast cell neoplasms: recommendations of the European Competence Network on Mastocytosis
Leukemia (2015)
-
Copy-neutral loss of heterozygosity and chromosome gains and losses are frequent in gastrointestinal stromal tumors
Molecular Cancer (2014)
-
Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumors: a meta-analysis
World Journal of Surgical Oncology (2014)
