
Abstract
The clinical outcome for osteosarcoma (OS) remains discouraging despite efforts to optimize treatment using conventional modalities including surgery, radiotherapy and chemotherapy. Novel therapeutic approaches based on our expanding understanding of the mechanisms of tumor cell killing have the potential to alter this situation. Tumor suppressor gene therapy aims to restore the function of a tumor suppressor gene lost or functionally inactivated in cancer cells. One such molecule, the p53 tumor suppressor gene plays a critical role in safeguarding the integrity of the genome and preventing tumorigenesis. Introduction of wild-type (wt) p53 into transformed cells has been shown to be lethal for most cancer cells in vitro, but clinical trials of p53 gene replacement have had limited success. Analysis of these clinical trials highlighted the insufficient efficacy of current vectors and low proapoptotic activity of wt p53 as a single agent in vivo. In this review, a contemporary summarization of the current status of adenovirus-mediated p53 gene therapy of OS is presented. Advancement in our understanding of p53 tumor suppressor activity, the molecular biology of chemoresistant OS, and recent advances in tumor targeting with adenoviral vectors are also addressed. Based on these parameters, prospects for future investigations are proposed.
Similar content being viewed by others


Main
Osteosarcoma (OS) is among the commonest of primary tumors of bone. It represents one of the most morphologically heterogeneous tumors known in human pathology and appears in distinct clinical forms with different degrees of malignancy.1 Most variants of OS are extremely aggressive as characterized by their rapid growth and early development of distant metastasis, mostly to lung and bones. Death from OS is usually the result of respiratory failure due to progressive pulmonary destruction from metastasis.2, 3
Standard treatment involves the use of ‘up-front’ multiagent chemotherapy, definitive surgery of the primary tumor, and postoperative chemotherapy. Introduction of adjuvant and neoadjuvant cytotoxic treatment and improvements in surgical procedures have increased 5-year survival to over 60% compared to < 20% when only surgery and/or radiation therapy had been used.4, 5 Despite attempts to further increase the disease-free survival for poorly responding patients through administration of more intensified therapy, no survival benefit has been convincingly shown.6, 7, 8
Gene therapy can be defined as the treatment of disease by the introduction of a therapeutic gene or the manipulation of a disease-related gene such as abrogation of an activated oncogene. Tumor suppressor gene therapy is that part of gene therapy which aims to restore the function of a tumor suppressor gene lost or functionally inactivated in cancer cells. In other words, tumor suppressor genes can be defined as genes for which loss-of-function mutations are oncogenic. Most commonly they regulate diverse cellular activities; cell-cycle checkpoint responses, detection and repair of DNA damage, protein ubiquitination and degradation, mitogenic signaling, cell specification, differentiation, migration and tumor angiogenesis.9 Many cancers have been shown to have inactivated tumor suppressor genes. The concept of tumor suppressor gene therapy is thus based on the following assumptions: (1) restoration of tumor suppressor function through gene transfer and expression would suppress the tumor phenotype of a cell and (2) normal nontransformed cells with functional endogenous tumor suppressor genes should tolerate the introduction of low to moderate levels of tumor suppressor genes.10 Indeed, reintroduction of known tumor suppressors into tumor cells in vitro has been shown to cause an acute change in cell physiology and gene expression, resulting in growth arrest or cell death.11 Despite the response of tumor suppressor gene introduction, the genotype of cancer cells remain markedly different from the putative normal counterparts from which they were derived due to quick acquisition of additional mutations after inactivation of tumor suppressor genes (genetic instability). Introduction of these genes in this context, in fact, cannot reverse the tumorigenic properties of such cells. Importantly, growth-arrested cancer cells are still metabolically active and can promote growth of other tumor cells in their vicinity.12, 13 Furthermore, some of these cells can also resume growing.14 Therefore, only those tumor suppressor genes which have the capacity to efficiently induce tumor cell death should be considered as good candidates for tumor suppressor gene therapy.
The p53 gene has an attractive apoptotic tumor suppressor profile as a gene therapy agent. Introduction of wild-type (wt) p53 causes death specifically in tumor cells, sparing normal cells. Additionally, p53 can promote death of nearby nontransduced tumor cells via its ability to transactivate genes whose products inhibit angiogenesis15 and via induction of an immune response against tumor cells,16 that is p53 causes a bystander effect. Results of preclinical studies on more than 100 cell lines and tissues have shown that introduction of wt p53 into neoplastic cells in vitro was lethal for most cells with p53mut or p53null genotypes.17 Unfortunately, clinical trials based on wt p53 gene replacement have had limited success.18, 19, 20 Furthermore, p53 gene therapy of metastatic disease did not result in improvement in patient status and survival.21 Analysis of these clinical trials revealed the insufficient efficacy of that generation of Ad vectors and low proapoptotic activity of wt p53 in vivo.22, 23, 24
This review presents a contemporary summarization of the current status of adenovirus-mediated p53 gene therapy of OS. We will first describe the recent advances in our understanding of the mechanisms of tumor suppressor activity of p53 and then focus our discussion on the use of Ad vector expressing p53 in the context of gene therapy of OS.
An overview of p53 tumor suppressor pathway
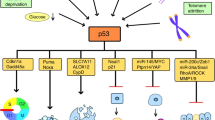
The particular contribution of p53 to certain functional decisions depends not only on p53. Depending on the plethora of intrinsic and extrinsic factors (the nature and intensity of stress, cellular and tissue contexts), activation of p53 results in different cells' fate.25, 26 Acting primarily as a transcription factor, p53 plays a critical role in regulating the cell cycle and maintaining genomic integrity by inducing growth arrest or apoptosis in response to a variety of stresses26, 27, 28, 29 (see Figure 1).

p53-mediated response to stress. A schematic diagram demonstrating some of the known components of the p53 functional circuit. Shown are key components of the apoptotic program as well as some its key regulators. Components in white squares inhibit apoptosis while those in gray squares promote apoptosis. See text for details.
The structure and function of p53: The p53 tumor suppressor gene encodes a 393 amino-acid modular protein with several interdependent functional domains (see Figure 2).30, 31 The acidic N-terminal domain (amino acids 1–63) is important for transactivation; it contains residues interacting with components of the basal transcriptional machinery.32 It is also critically involved in regulating the stability and activity of p53 protein via interactions with Mdm2.33, 34 A region in this domain was identified (amino acids 43–63) as being required for p53-dependent apoptosis.35, 36 The proline-rich region (amino acids 64–91) is also necessary for apoptosis.37, 38, 39 The sequence-specific DNA-binding domain within residues 100–300 serves as the core of the p53 protein. The majority of missense mutations found in tumor samples occurs in this region of the gene and result in disruption of the ability of p53 to specifically bind DNA. The oligomerization domain of p53 (amino acids 324–355) participates in the formation of p53 tetramers. The C-terminal basic domain (amino acids 356–393) is an important regulatory domain. One of its functions is to keep p53 in an inactive form until modifications including phosphorylation and acetylation can serve to activate the protein allosterically.30

Functional domains of p53 protein. The human p53 protein consists of 393 amino acids and has several functional domains (indicated by the shaded regions) including an acidic or transactivation domain, a proline-rich domain, a sequence-specific DNA-binding domain, an oligomerization domain, and a C-terminal regulatory domain. See text for details.
In unstressed cells, p53 is present at low levels and exists in a latent form that requires modification to become active. Various forms of stress (DNA damage, oncogenic stimuli, redox stress and hypoxia) rapidly induce a transient increase in p53 protein via enhanced translation of p53 mRNA and stabilization of p53 protein.40 Also, a series of post-translational modifications occurs in p53 including phosphorylation, glycosylation and acetylation, that affect a myriad of activities including protein–protein interactions, alternative splicing, homo- and hetero-oligomerization, C- and N-terminal truncations, and DNA-binding capabilities.30, 40 Activated p53 initiates a transcriptional program that reflects the nature of the stress signal, resulting in protein modifications and alternative proteins being associated with p53 protein.41
The function of p53 in cancers can be lost by various mechanisms, including lesions that prevent activation of p53, mutations within the TP53 gene (which encodes p53) itself and mutations of downstream mediators of p53 function. Most TP53 mutations found in tumors are point mutations which usually result in the expression of mutant p53 protein. Importantly, these proteins are often more stable than wt p53 and some p53 mutants can act as dominant-negative inhibitors of wt p53.42, 43 Certain missense mutations not only result in abrogation of wt p53 activity, but the expressed p53 mutant proteins also tend to gain oncogenic function, such as interference with wt p53-independent apoptosis.44 Importantly, such p53 mutants contribute to the resistance of tumor cells to commonly used chemotherapeutic agents.45 Many tumors with a dysfunctional p53 pathway retain wt protein. Defects of the tumor suppressor pathway in these cases results from functional abnormalities in upstream regulators of p53 activity or from effectors of apoptotic activity of p5346 (see Figure 1).
Mdm2
Mdm2 is a master regulator of p53.47 It controls p53 function through inhibition of p53-mediated transcriptional activity48, 49 and by promoting p53 degradation.33, 34 Binding of Mdm2 to p53 is essential for this effect. Importantly, the mdm2 gene is a target gene for p53. p53 binds to p53-responsive elements located within the mdm2 gene, and promotes the production of mdm2 transcripts.50, 51 Consequently, an autoregulatory negative feedback loop exists, wherein p53 induces Mdm2 expression and Mdm2 represses p53 activity. This serves as an important mechanism for restraining p53 function in the absence of stress. Cellular stress induces modifications of both p53 and Mdm2 proteins, which results in reduced avidity of p53 for Mdm2 and thereby, inhibition of ubiquitination and degradation of p53 protein. An inappropriate excess of Mdm2 may result in exaggerated silencing of p53, abrogating its tumor suppressor effects, as it was found that overexpression of Mdm2 in mice resulted in the production of tumors with as little as a four-fold increase in Mdm2 levels giving rise to tumors in 100% of the mice.52
Multiple mechanisms may modulate Mdm2 activity and its ability to access p53 for ubiquitination. For example, p14ARF, human ribosomal protein L11, and cyclin G can act as negative regulators of Mdm2's ability to downregulate p53 levels.53, 54, 55 Other factors recently shown to positively regulate Mdm2 include YY1, gankyrin and KAP1.56, 57, 58 Mdm2 and mdmx prevent ASPP1 and ASPP2 from stimulating the apoptotic function of p53 by binding and inhibiting the transcriptional activity of p53.59
Proapoptotic activity of p53
P53 can induce apoptosis in several ways.60, 61, 62, 63 It can act as a transcriptional factor that directly activates the expression of genes known to promote apoptosis including Bax,64 Bak,65 PUMA,66, 67 Noxa68 and Bid.69 The net effect of induction of these genes is an increasing of the ratio of pro- to antiapoptotic Bcl-2 proteins, consequently, favoring the release of apoptogenic proteins from the mitochondria, caspase activation and apoptosis. In addition, p53 can also transactivate several effectors of apoptosis acting downstream of mitochondria such as Apaf-1 and caspase-6.70, 71 p53 also regulates the extrinsic apoptotic pathway, activating expression of Fas/CD95, DR5 and RDL1 genes72, 73 and is capable of counteracting with antiapoptotic pathways inducing genes including PTEN and IGF-BP3, which inhibit survival signaling.74, 75
P53 also possesses a proapoptotic function that is independent of its transcriptional activity.76, 77, 78 In response to stress, p53 accumulates in the cytoplasm, where it directly or indirectly activates the proapoptotic proteins to promote mitochondrial outer-membrane permeabilization.79, 80, 81, 82 It seems likely that coordination of the nuclear, cytoplasmic, and mitochondrial activities of p53 cooperate to ultimately cause cell death. In addition to transactivation functions, p53 has also transrepression capabilities that may contribute to apoptosis.83, 84, 85
To explain how p53 can induce apoptosis or cell-cycle arrest, different models have been proposed. One quantitative model assumes the existence of p53-responsive genes with differing binding affinities. According to this model, a subset of promoters should be activated only when the expression level of p53 reaches a certain threshold. Low levels of p53 protein will result in cell-cycle arrest because promoters of cell-cycle arrest genes (p21, mdm2) have higher affinity binding sites for p53 than promoters of genes responsive for apoptosis (Bax, IGF-BP3).36 Not all data support this model. For example, the binding affinity of p53 to the PUMA promoter, which regulates a proapoptotic gene, is similar to the p21 and mdm2 promoters. Other qualitative models proposed that induction of apoptosis or cell-cycle arrest by p53 depends on p53-binding proteins, which are able to modulate the selection of target genes. Importantly, some of these factors have a specific and selective role in enabling the expression of the apoptotic target genes. For example, JMY cooperates with p300 to enhance the ability of p53 to induce the expression of BAX, but does not significantly influence the induction of the p21 gene.86 The ASPP1 and ASPP2 genes have been shown to interact with p53 and enhance the interaction with the promoter of the BAX gene.87 Inhibition of ASPP expression blocks the apoptotic response to p53. Other important modulators of p53 apoptotic induction are p63 and p73 proteins. It was shown that p53 is not able to bind promoters of apoptotic targets in p63/p73 double-null cells.88
E2F
The E2F family of transcription factors are downstream effectors of the retinoblastoma (Rb) protein pathway. Since their discovery, E2Fs have been viewed as positive regulators of genes required for DNA synthesis.89, 90 Recent findings have changed this view.91, 92, 93 It has been found that depending on the context, E2Fs function as activators or repressors of transcription. Specifically, there is a bifurcation of E2F family members with respect to their transcriptional roles in cell-cycle control; E2F1–3 function mainly as activators of transcription while E2F4 and E2F5 act primarily as repressors. These, new findings considerably broadened our understanding of E2F transcriptional control beyond S phase, E2F also regulates genes involved in DNA repair and recombination, apoptosis, differentiation and development, as well as other genes of still unknown function.
E2F can induce apoptosis in a p53-dependent manner as well as in a p53-independent manner.92, 94 E2F1 transcriptionally regulates the expression of the DNA damage-responsive kinases ATM and Chk2 that phosphorylate p53,95, 96 and induces expression of the proapoptotic cofactors of p53, ASPP1, ASPP2, JMY and TP53INP1.97, 98 E2F also directly activates the p53 proapoptotic target genes PUMA, Noxa, Bim, Hrk/DR5,99 Apaf170 and SIVA,100 and downregulates the expression of Mcl-1, an antiapoptotic member of the Bcl-2 family.92
E2F factors have been suggested to potentially be involved in acquired resistance to chemotherapeutic drugs in OS cells. Recently, it has been found that E2F1-mediated activation of the p73 promoter induces apoptosis in human OS cells.101 Cinti and colleagues have also demonstrated that doxorubicin treatment of chemosensitive human OS cells (HOS) results in increased levels of p73, overexpression of bax, and downregulation of bcl-2. In performing a chromatin immunoprecipitation assay, they detected a multimolecular complex formed by E2F1-pRb2/p130–p300 bound to the p73 promoter.102 In contrast, in chemoresistant cells (HOSDXR150) a multimolecular complex formed by E2F4-pRb2/p130-histone deacetylase complex 1 was bound to the same p73 promoter region. In another study, significant correlations between E2F and dihydrofolate reductase and thymidilate synthase (TS) in OS patient samples were observed, suggesting the potential involvement of the E2F pathway in methotrexat resistance.103
PTEN
In recent years, it has been demonstrated that multiple levels of crosstalk exist between PTEN (phosphates and tensin homolog deleted from chromosome 10) and p53, and that functional PTEN is important for proapoptotic activity of p53. The PTEN tumor suppressor gene is located on chromosome 10q23 and is known to be the most highly mutated tumor suppressor gene after p53.104 The PTEN encodes a dual specificity protein–lipid phosphatase that plays an important role in regulating proliferation, migration, survival, cell invasion and tumor angiogenesis.105, 106 The lipid phosphatase activity of PTEN is crucial for its tumor suppressive activity. The main PTEN substrate is phosphatydilinositol(3,4,5)-triphosphate (PIP-3). PIP-3 levels are increased upon stimulation by growth factors through activation of PI3 kinase. Accumulation of PIP3 at the membrane allows recruitment of the proto-oncogene serine/threonine kinase Akt/PKB which is then activated by phosphorylation. Activated Akt is a well-known survival factor. Thus, by keeping the levels of PIP-3 low, PTEN prevents prosurvival activity of Akt. Akt also phosphorylates serine residues in the Mdm2 domain that contains a nuclear localization motif. These phosphorylations are important for translocation of Mdm2 from the cytoplasm into the nucleus where it can interact with p53 and inhibit its transcriptional activity.107, 108 Consequently, PTEN protects p53 from survival signaling induced by growth factors via the PI3K-Akt pathway and nuclear entry of Mdm2. Recently Wu and colleagues have shown that by antagonizing the PI3-kinase pathway, PTEN also negatively regulates the P1 promoter on the 5′ end of the mdm2 gene, leading to decreased L-Mdm2 expression and reduced p90MDM2 isoform production.109
p53 is capable of binding directly to a site in the PTEN promoter and inducing PTEN mRNA expression in response to DNA damage.75 In turn, a p53-binding domain has been identified in PTEN and evidence exists that physical interaction between PTEN and p53 is important for modulation of p53 function in vivo.110, 111 It was further demonstrated that PTEN can control p53 half-life independent of PI3K/Akt/Mdm2 via a currently unknown mechanism. Finally, Donner and colleagues have recently demonstrated that intensive stress induces the phosphorylation of p53 protein which results in a switch of gene promoter targeting; from the promoter for mdm2 to the promoter for PTEN. Formation of the mdm2-p53 autoregulatory feedback loop is thus blocked and p53 together with PTEN form an apoptotic amplification cycle that induces cell death.107 These findings show the importance of cooperation of PTEN with p53 for efficient action as a tumor suppressor and provide a strong rationale for combined p53+PTEN cancer gene therapy.
Despite an overall high incidence of PTEN mutations in humans, the majority of them have been identified in epithelial cell-derived tumors.112 To date, PTEN mutations have not been associated with OS tumorigenesis, despite the fact that the chromosomal loss of 10q has been documented in nearly 30% of human OS tumor samples analyzed.113
Prosurvival (antiapoptotic) signaling in tumor cells
Survival signaling provided by the tumor microenvironment significantly modulates cellular response to p53 activation. Interactions of cancer cells with each other and with stromal cells, various growth factors and with components of the extracellular matrix can drastically affect the apoptosis sensitivity of these cells.114, 115
One of the most important molecules responsible for rescuing cells from p53-induced apoptosis is Akt kinase.116 The activation of Akt by survival signals is achieved mainly through a kinase cascade involving PI3 kinase. The mechanism by which Akt protects cells from death is multifactorial.117 Activated Akt kinase can phosphorylate and downregulate expression of PUMA, an important mediator of the p53 apoptotic response and also can inhibit catalytic activity of caspase-9.118 Akt-mediated phosphorylation of FKHR—a member of the Forkhead family of transcription factors, prevents its nuclear translocation and activation of FKHR target genes, which include the proapoptotic proteins BIM and Fas ligand.119 Also, the apoptosis-inducing protein Bad after phosphorylation by Akt loses the ability to bind antiapoptotic members of the Bcl-2 family, specifically Bcl-2 and Bcl-XL, thus releasing them for a cell survival response.120 As was alluded to previously, Akt-mediated phosphorylation of Mdm2 results in inactivation of p53.
Experiments conducted using three-dimensional (3D) in vitro models suggest that additional levels of apoptosis signaling regulation likely exist in tissues.121, 122, 123 Furthermore, it has been shown that fundamental differences exist between degrees of survival of tumor cells incorporated into 3D spheroids vs tumors grown in monolayer in response to exposure to death stimuli.124 Cells in 3D spheroids rapidly acquire multidrug resistance and this phenotype appears to be linked to cell adhesion.
Inhibition of tumor angiogenesis and metastases
The p53 protein stimulates the expression of genes that prevent the growth of new blood vessels in the tumor's vicinity and inhibits tumor invasion and metastasis. Among known p53's target genes with antiangiogenic and antimetastatic activities are thrombospondin-1 (TSP1), KAI1, BAI1 and maspin.125, 126, 127, 128 Reintroduction of wt p53 into tumor cells upregulated the expression of antiangiogenic factors which leads to downregulation of proangiogenic vascular endothelial growth factor and inhibition of neovascularization.129, 130, 131, 132, 133 Suppression of angiogenesis contributed to suppression of both primary tumors and their metastasis.134
Role of the immune system in p53 tumor suppressor gene therapy
Despite the fact that the immune system has a huge antitumor potential, tumors are able to escape from immunosurveillance. Rational modulation of the patient's immune response could significantly augment the antitumor efficacy of gene therapy strategies. Interestingly, a link was recently discovered between p53 and the interferon system in modulating tumor suppression and antiviral immunity. The interferon system can upregulate expression of the p53 gene which in the context of other stimuli leads to p53 activation and enhancement of its proapoptotic activity.16, 135 In turn, p53 protein can transactivate expression of the interferon regulatory factor 5 gene,136 whose product contributes to IFN-α and IFN-β induction.137 The type I interferons exhibit a wide range of biological activities including antiviral, antiproliferative and antiangiogenic effects.138, 139 They activate cells in the immune system, and help in presentation of cancer cells to the immune system by stimulating production of cell surface molecules (costimulatory molecules, MHC antigens and tumor-associated antigens).
When p53 was overexpressed in the context of insertion of a viral vector in tumor cells, it was seen to induce an antitumor immune response.140, 141, 142, 143 This response was mediated by CD4+ Th cells, CD8+ cytolytic T cells and NK cells and resulted not only in tumor cell rejection but also in establishing long-term immune memory.144 Importantly, rejection of poorly immunogenic tumors and well-established progressing tumors has been demonstrated when introduction of p53 was combined with inhibition of immunosuppressive activity of Treg cells and activation of innate immunity.145 Particularly important, this tumor rejection occurred in the absence of autoreactivity to normal tissues.145, 146
Thus, p53 inhibits tumor progression not only via induction of apoptotic cell death (an ‘intrinsic’ tumor suppressor mechanism), but also via stimulation of antitumor immunity (an ‘extrinsic’ tumor suppressor mechanism). This is important because reintroduction of p53 into every cell in a solid tumor is not currently possible. Induction of an efficient antitumor immune response should help to eliminate uninfected tumor cells and prevent relapse. Nevertheless, as a high ratio of activated immune cells to tumor cells is required for immunologic tumor destruction,147 it is important to eliminate (kill) as many tumor cells as possible by other means. In this regard the oncolytic capabilities of a conditionally replicating adenovirus (CRAd) agent could be advantageous for this purpose.
p53 in development and differentiation
In addition to its role as a tumor suppressor and cell-cycle checkpoint control protein, p53 has been implicated as an important protein in development and differentiation.148, 149, 150, 151 Particularly interesting are the findings regarding p53's role in stem cell self-renewal and differentiation. It has been shown that p53 induces differentiation of mouse embryonic stem cells (ESC).152 This role of p53 in cellular differentiation represents an alternative mechanism to maintaining genetic stability in ESCs, which are resistant to p53-dependent cell-cycle arrest and apoptosis after DNA damage.153 Induction of the differentiation of ESCs into cell types that can undergo efficient p53-dependent cell-cycle arrest or apoptosis will result in the elimination of genetically abnormal cells. p53 also regulates asymmetric self-renewal and immortal DNA strand cosegregation—two unique attributes of adult stem cells (ASC), which have been proposed to restrict their carcinogenic potential.154 Asymmetric self-renewal is characterized by divisions that produce a new ASC and a nonstem cell sister. Immortal DNA strand cosegregation means that at each cell division, stem cells continuously nonrandomly segregate to themselves the set of chromosomes with the oldest template DNA strands (ie, immortal DNA strands). This molecular maneuver allows ASC to significantly reduce the number of mutations which arise from unrepaired or misrepaired replication errors. Using engineered cultured cells that exhibit asymmetric self-renewal and immortal DNA strand cosegregation, Sherley and colleagues have shown that both ASC-specific functions are regulated by the p53 gene.154 Recently, two groups demonstrated increased self-renewal of neural stem cells in mice lacking p53.155, 156 Prenatal exposure of these mice to mutagens lead to the formation of glioblastoma-like tumor in the adult supraventricular zone of the cerebrum.155
Role of p53 in the development of bone and in OS pathogenesis
Bone malformations (failures in skull growth and delayed longitudinal bone growth) and a high incidence of OSs in p53 null or mutant mice suggest that p53's function may be critical during skeletogenesis.155, 157, 158 Abnormalities of the p53 gene in OS occur with frequencies approaching 50% of all cases.159 In in vitro studies it has been shown that p53 loss of function mutations often lead to a lack of terminal differentiation for the osteogenic lineage, and this property is regained after stable transfection of the cells with wt p53.160 Analysis of clonal mesenchymal stem cell (MSC) populations derived from p53−/− mice have revealed that Runx2 and osteopontin, early and intermediate osteogenic markers, were upregulated in p53−/− MSC compared to wt cells during osteogenesis.161 Interestingly, the terminal osteogenic marker gene, osteocalcin was lower in p53−/− MSC. Jones and colleagues have also found that p53-null osteoprogenitor cells have increased expression of Runx2. In contrast, expression of Runx2 has been lost in osteoblasts with hyperactive p53.162 Wang et al have reported that osterix, another transcription factor regulating bone formation and osteoblast differentiation,163 was upregulated in calvarial bones of p53−/− mice.164 Thus, it seems, that the lack of p53 function results in enhanced early osteogenesis in MSC, but prevents terminal differentiation toward a mature osteocyte phenotype.
Particularly interesting are findings that p53-null osteoprogenitor cells have increased proliferation, increased expression of Runx2, and increased tumorigenic potential, as mice specifically p53 deleted develop OS.162 Runx2 belongs to the RUNX (Runt-related transcription factor) family of genes which encode transcription factors that can bind and recruit a range of coactivators or corepressors (CoR) and thereby serve as orchestrators of transcription at target promoter sites.165, 166 Runx2 promotes skeletal cell differentiation from multipotential mesenchymal cells,167, 168, 169 and also contributes to cell growth control of osteogenic cells.170, 171 Its expression is tightly regulated by a broad spectrum of factors including (1, 25)-dihydroxyvitamin D3, ascorbic acid, bone morphogenetic protein-2 (BMP-2), transforming growth factor-β, tumor necrosis factor-α, fibroblast growth factor-2, retinoic acid and parathyroid hormone.172 p53 functionally interact with many of these factors and lack of p53 might explain the fact that the control of Runx2 levels and its function is consistently disrupted in OS cells.171, 173 For example, p53-deficient cells display an impaired cytostatic response to TGF-β signals.174 Also, it has been shown that Runx2 induces the growth arrest of osteoblastic cells through induction of p27KIP1.173 This function of Runx2 might be inefficient in p53-null cells, because p27 acts as a haploinsufficient tumor suppressor,175 but it has been shown that downregulation of p27 is a common feature in p53−/− tumors.176 From the point of view of OS pathogenesis, particularly interesting is the realization that Runx2 has a survival function,177 and due to this ability, probably collaborates with other oncogenes in tumor development.178 As will be discussed below, deregulation of Wnt signaling pathway in p53−/− osteoblasts appears to result in the upregulation of c-myc.
Presence of functional p53 is important for regulated expression of other factors involved in osteogenesis especially osteocalcin179 and BMP-2.180 Noted too are the findings about interactions between p53 and the Wnt/β-catenin signaling pathway in osteoblasts181 in light of the emerging role of the Wnt pathway in controlling osteoblast differentiation.182, 183, 184, 185, 186 The Wnt/β-catenin signaling pathway is transduced through stabilizing β-catenin protein by inhibiting glycogen synthase kinase-3-mediated β-catenin phosphorylation.187 Unphosphorylated β-catenin accumulates in the cytoplasm and translocates to the nucleus where it displaces CoR from the Lef1/Tcf transcription factor. This, in turn, directly interacts with Lef1/Tcf and recruits transcriptional coactivators to stimulate expression of many genes, including c-myc and cyclin D1. Approximately 70% of OS samples displayed deregulation of β-catenin.188
Recently, it has been demonstrated that the inactivation of p53 in OS directly by mutation vs indirectly by human HDM2 amplification may have different cellular consequences with respect to the stability of the genome; mutations of p53 correlated significantly with the presence of high levels of genomic instability while inhibition of tumor suppressive properties of p53, via amplification and overexpression of HDM2, did not displayed similar high levels.189 Also, mice bearing certain ‘knock-in’ p53 mutants have a tendency for development of metastasizing OS.190, 191 Studying the properties of such TP53 mutants has demonstrated their ability to bind and inhibit the function of p63 and p73. Notably, these data indicate in vivo differences between loss of p53 or presence of a p53 mutation in tumor development and have significant implications for therapy.191
Molecular profiling of chemoresistant OS
Gene expression profiling of chemotherapy-resistant OS has been recently explored using microarray technology.192, 193, 194 These studies revealed that the chemoresistance signature includes genes related to ‘bone metabolism,’ ‘cancer biology’ and ‘drug resistance’. Particularly important are the findings regarding dysregulation of TWIST1 overexpression in chemoresistant OS cells.192 TWIST1 has been shown to affect the expression and DNA-binding ability of Runx2.195, 196 In addition, TWIST1 has been found to cooperate with MYC and MYCN to suppress the p53-dependent apoptosis pathway,197, 198 and is involved in Taxol resistance and metastasis.199, 200 Importantly, overexpression of TWIST1 is associated with characteristics of osteoprogenitor cells that have decreased proliferation and a less mature phenotype.201 As all of these are properties of cancer stem cells,202, 203 Lau and colleagues have suggested that chemoresistant OS cells overexpressing TWIST1 may arise from putative cancer stem cells.192 Cells with stem cell qualities have been identified in malignancies of hematopoietic origin and in some solid tumors.202 According to current understanding, cancer stem cells are likely to share many of the properties of normal stem cells, including relative quiescence, resistance to drug and toxins through the expression of ABC transporters, capacity for DNA repair, and resistance to apoptosis.202 These features combined with the ability for unlimited self-renewal might explain the failure of current therapies to eradicate solid tumors. Thus, in order to cure cancer, anticancer agents must be able to kill cancer stem cells.
p53 tumor suppressor gene therapy of OS
Up until very recently only one cell line, Saos-2, or its derivatives were used in tumor suppressor gene therapy studies of OS.204, 205, 206, 207, 208 This cell line has a homozygous deletion of the p53 gene and also lacks expression of the Rb gene. Reintroduction of wt p53 in Saos-2 induces cell death by apoptosis.205 Importantly, when delivered and expressed efficiently using Ad vectors, wt p53 alone induces apoptotic death of Saos-2 cells without additional stimuli. Sensitivity to p53 proapoptotic activity therefore makes Saos-2 cells a suitable system for the analysis of certain aspects of tumor suppressor gene therapy. For example, Densmore et al have advantageously employed the Saos-2 cell system to study the potential of polyethyleneimine as a p53 gene delivery vehicle to OS lung metastases. Using the Saos-LM6 cell line, a metastatic derivative of the Saos-2 line, they demonstrated that aerosol delivery of plasmid DNA/polyethylenemine complexes containing either wt p53 gene or a constitutively active p53-CD(1–366) to mice with established lung micrometastases resulted in significant reduction in the numbers and size of tumors.209 In another study Song and Boyce used Saos-2 cells to explore the use of an insect baculovirus as a vector for gene delivery.210 They showed that infection with this vector expressing wt p53 (BV-p53) at a MOI of 100 (at which nearly 100% of cells were transduced) resulted in apoptotic cell death in about 50% of cells. When the same dose of BV-p53 was used in combination with adriamycin (a DNA-damaging chemotherapeutic drug), >95% of Saos-2 cells were killed.
Despite demonstrated suitability of Saos-2 cells for studying certain questions of OS biology, a wide panel of OS tumor cell lines would be preferable to provide more reliable information regarding the feasibility of tumor suppressor gene therapy. In this respect, in recent studies by Ganjavi et al211 and Hellwinkel et al212 attempts were made to explore the antitumor efficacy of tumor suppressor gene reintroduction using a panel of different human OS cell lines. Ganjavi and associates used HOS, KHOS/NP and MNNG cell lines in addition to Saos-2 cells. It should be noted that KHOS/NP and MNNG are derivatives of HOS cells. In these studies infection with Ad-p53wt at an MOI of 8 p.f.u./cell (16 p.f.u./cell for KHOS/NP) induced apoptosis in approximately half the cell population in each cell line. Introduction of p53 in these cell lines dramatically enhanced their sensitivity to DNA damaging drugs cisplatin and doxorubicin.
Hellwinkel et al used a panel of five human OS cell lines. Results of these studies demonstrated substantial cell death only in two of five tumor cell lines tested. Again, Saos-2 cells demonstrated highest sensitivity to wt p53; only 14% of these cells were alive after infection with Ad-p53wt at an MOI of 25. U2OS cells have also demonstrated sensitivity to wt p53 reintroduction, but a much higher MOI was required to induce cell death. Approximately 70% of U2OS cells were alive after infection with Ad-p53wt at an MOI of 25, and MOIs of 100 and 200 were required to achieve 90% cell killing. It is important to note that Ad5-based adenoviral vector transduces U2OS cells more efficiently than Saos-2 cells. The lower response of U2OS cells to wt p53 introduction might be explained by inhibition of p53 activity by Mdm2 which is overexpressed in U2OS cells. Three other OS cell lines studied (K-HOS, MG-63 and SJSA) have demonstrated diminished cell proliferation in response to Ad-p53 infection but not cell death. The results with SJSA and MG-63 cells can be explained by low efficiency of Ad transduction of these cells and overexpression of Mdm2.213, 214, 215, 216 The reasons for the very low apoptotic response of K-HOS cells on p53 introduction remain unknown.
To explore whether genotoxic stress could augment the response of studied OS cells to p53 introduction, Hellwinkel et al combined Ad-p53 infection with treatment by cisplatin or doxorubicin. Except for U2OS and Saos-2 cells, the drugs did not multiply the effects of p53 introduction. Further results by this group demonstrated that efficient induction of apoptosis in OS cells, via introduction of a wt p53 tumor suppressor gene using an Ad5 based adenoviral vector, can be achieved only in cases where OS cells express high levels of Ad5 receptors and do not express p53 inhibitors.
About 30% of OS tumors overexpress Mdm2 due to mdm2 gene amplification,213 and, consequently, introduction of wt p53 is unlikely to be efficacious. To overcome the inhibition of p53 by Mdm2, modified p53 genes have been constructed. Specifically, Conseiller et al217 generated a chimeric tumor suppressor 1 (CTS1) containing the p53 core domain (amino acids 75–325), with the VP16 activation domain ligated to the 5′-end. They further optimized the leucine-zipper domain that was ligated to the 3′-end of the p53 core domain. This CTS1 demonstrated resistance to inactivation by Mdm2 and by the p53 mutant, H175. In a transient transfection assay with Saos-2 cells, CTS1 has shown the ability to induce apoptosis faster than wt p53 protein. To explore the antitumor potential of CTS1 in the context of Mdm2 overexpression, Bougeret et al218 constructed an Ad vector expressing CTS1 under control of the CMV promoter (AV.CTS1). Using the human OS cell line SA1 (also known as Osa-CL) which overexpresses endogenous Mdm2, they found that AV.CTS1 induced apoptosis six-fold more efficiently than Ad-expressing wt p53 (Ad-p53) in vitro. Furthermore, induction of apoptosis was significantly higher in AV.CTS1-transduced pre-established SA1 tumors in nude mice compared to Ad-p53-treated tumors. In contrast, AV.CTS1 induced cell-cycle arrest and cell death less efficiently than Ad-p53 in tumor cells harboring nonfunctional p53. Results of studies with CTS1 have shown that modified p53 can be more efficient than wt p53 in induction of apoptosis in cells with high levels of Mdm2.
A different approach was used by Tang et al219 to overcome the inhibition of p53 by Mdm2. Previous work had established that the hydrophobic residues Leu-14 and Phe-19 of p53 were crucial for the interaction between p53 and Hdm2.32 To study proapoptotic properties of p53 with amino-acid substitutions at Leu-14 and Phe-19, Lin and colleagues infected cells with different levels of Mdm2 with an adenoviral vector expressing a modified p53 gene (Ad-p53 14/19).219 They found that in HT1080 cells (low Mdm2), Ad-p53 14/19 induced apoptosis at a level similar to Ad-wtp53. In human OS SJSA cells (high Mdm2) Ad-p53 14/19 induced dramatically more apoptosis than Ad-wtp53 (44 vs 11%). Also, infection of SJSA cells with Ad-p53 14/19 sensitized them to doxorubicin and cisplatin. After exposure of SJSA cells to 0.1 μg ml−1 of doxorubicin or 5 μg ml−1 cisplatin only 4 and 24%, respectively, were apoptotic. Combined treatment of SJSA cells with Ad-wtp53 plus doxorubicin or cisplatin at the same doses induced apoptotic cell death only in 15 or 33% of cells, respectively, demonstrating continued resistance. In contrast, Ad-p53 14/19 plus doxorubicin or cisplatin generated apoptosis in 63 or 80% of SJSA, respectively. Thus, the studies of Bougeret et al and Tang et al demonstrated that abrogation of the Mdm2 binding site in p53 results in augmentation of its proapoptotic activity in OS cells with a high level of Mdm2 expression comparing to wt p53.
p53 in the context of CRAd agents (armed CRAds)
The anticancer potency of p53 in the context of a nonreplicative Ad vector is limited because the vector infects only a small fraction of the cells within a tumor. Employment of a replication-competent Ad vector has the potential of transgene delivery to significantly more cells via repeated cycles of infection, cell lysis, and virus spread to neighboring cells.220, 221 Also, replication-competent Ad vectors have the potential to increase transgene expression up to 1000-fold in each infected cell, compared with a nonreplicating Ad. However, p53 expression might be deleterious for viral replication due to induction of preliminary apoptosis. Also, spreading of the Ad vector through the tumoral mass might be blocked due to induction of the interferon system by p53.16 Alternatively, several Ad5 early gene products oppose p53 function. For example, adenoviral E1B-55-kDa protein binds and inactivates p53.222 The E1B-55-kDa protein and another Ad protein E4orf6 together mediate the ubiquitination and degradation of p53.223, 224, 225, 226 Other Ad proteins can indirectly interfere with p53 activity. The E1A gene products antagonize p53 activity and this correlates with its ability to interact with p300 and the p400/TRRAP protein complex.227, 228, 229 The E1B-19-kDa protein inhibits the downstream effects of p53 by blocking induction of apoptosis.230, 231 In contrast, the Ad5 E3 region encodes the adenoviral death protein which is cytotoxic but, owing to strong expression only at very late stages of viral infection, is not deleterious for viral production and promotes lysis of infected cells.232
Therefore, expression of p53 at late stages of the viral life cycle might be not deleterious to virion progeny production. To explore this strategy, Sauthoff et al233 constructed a replicating Ad vector (Adp53rc) that contains the p53 cDNA inserted into the adenoviral fiber transcription unit, using an internal ribosomal entry site and demonstrated that the expression pattern of the p53 protein correlated with the late expression pattern of the adenoviral fiber protein. Adp53rc demonstrated improved oncolytic properties compared to the control Ad vector (Ad-co) which had the same genetic backbone, but lacked the p53 gene. Despite this, Adp53rc induced apoptotic cell death only in 10% of infected cells, compared with 5% induced by infection with Ad-co. Also, expression of the p53 target genes p21 and bax were not detected despite strong nuclear expression of p53. Thus, p53 caused no obstacles to viral replication but p53's properties as a transcriptional activator were significantly inhibited by Ad.
It is possible that despite the Ad genes which oppose p53 functions (which are strongly expressed at early stages of viral replication) low concentrations of these proteins might be sufficient to inhibit p53-dependent transactivation at late stages of infection. The E1B-55-kDA protein deserves special attention in this regard. It has several activities that inhibit p53 function.234 According to this model E1B-55K inhibits activation by p53 by binding a repression domain to it. Importantly, E1B-55K increases p53 stability by binding to the same region in the amino-terminal domain of p53 bound by Mdm2.32 Also, p53 bound to E1B-55K has a 10-fold higher affinity for its binding site than free p53.235 As a result, p53 is converted from an activator into a constitutive repressor of genes with p53-binding sites. The stabilization of p53 by E1B-55-kDa protein in the absence of E4orf6 protein leads to a significant increase in p53 concentration.236 Thus, E1B-55K will significantly limit p53 tumor suppressor activity in the context of replication competent Ad vector. To prevent such inhibition, modified p53 might be used instead of wt p53. For example, Dobbelstein and colleagues found that replacement of the five amino acid residues at position 24–28 by the homologous residues from p73 resulted in complete resistance of chimeric p53 (p53mt24–28) to E1B-55K- and E4orf6-mediated degradation.225 This chimeric p53 when expressed in the context of a first-generation Ad vector demonstrated resistance to degradation by Ad proteins in cells coinfected with wt Ad.237 Importantly, chimeric p53 was capable of inducing such p53 target genes as p21/CDKN1A, mdm2 and bax, regardless of the infection with replicative viruses.237, 238 Interesting, even strong overexpression of p53mt24–28 allowed the virus to replicate as efficiently as when it was absent. These findings demonstrate that employment of modified p53 in the context of a replication-competent Ad vector is a feasible approach.
The efficiency of p53 in the context of CRAd agents on OS cell lines has been studied by Gerritsen and colleagues.215, 239 To restrict Ad replication to tumor cells, amino acids 122–129 in the conserved region 2 domain of E1A (necessary for binding to the Rb protein) were deleted. Using a crystal violet assay, van Beusechem et al found that a CRAd agent expressing wt p53 (AdΔ24-p53) exhibited enhanced oncolytic capacity compared with a control vector (AdΔ24) in killing OS cells with low levels of Mdm2. AdΔ24-p53 was ∼100-fold more potent than AdΔ24 against U2OS cells and 10-fold more potent against Saos-2 cells, but had no advantage when examined in MG-63 and MNNG-HOS cells, which express high levels of Mdm2. In contrast, a CRAd agent expressing a p53 variant resistant to Mdm2-mediated degradation (AdΔ24-p53(14/19)) was ∼10 times more effective than either AdΔ24 or AdΔ24-p53 in killing Mdm2-high cells. However, AdΔ24-p53 (14/19) was less effective than AdΔ24-p53 against Mdm2-low cells. It should be noted that the low Coxsackie and Adenovirus Receptor (CAR) and high-Mdm2 MG-63 OS cell line was highly resistant to all CRAd agents tested and required 100 p.f.u./cell or more of AdΔ24-p53 (14/19) to be eradicated. Thus, significant improvements in CRAd agents are needed to efficiently eliminate OS tumors with high levels of Mdm2. In this regard, it seems that a p53 variant resistant only to Mdm2 is not an optimal alternative to p53 in a CRAd context. As E1B-55-kDa protein binds residues 23–27 of p53,32 p53(14/19) is still a candidate construct for inhibition and degradation. It is tempting to speculate that substitution of amino-acid residues 14 and 19 of p53 combined with replacement of residues at position 24–28 by the homologous residues from p73 might result in resistance of chimeric p53 to degradation by Mdm2 and Ad proteins. Such modified proteins should still retain the capability to transactivate p53-responsive genes. Also, capsid modification of Ad vector to target OS tumors expressing low levels of CAR receptors should enhance the oncolytic potential of CRAd agents.
Enhancement of transduction efficiency of OS cells with Ad vectors
Adenoviral vectors often transduce tumor cells inefficiently due to low levels of adenovirus receptor molecules on these cells. The adenovirus enters cells in two separate steps. First, the knob domain of the adenovirus fiber binds the primary attachment receptor. Specifically, the Ad5 and Ad2 viruses whose genetic backbone is mostly used for adenoviral vector construction bind to the CAR.240 The second step is internalization into the target cell which is mediated by an interaction of the Ad virion with avb3 and avb5 integrins.241 The transduction efficiency of Ad is known to correlate with levels of primary and secondary receptors on the cell surface.242, 243 It has thus been recognized that improved tumor cell transduction could be achieved via redirection of the adenoviral vector to non-CAR cellular entry pathways in CAR-deficient neoplastic cells.244, 245 This understanding has resulted in the generation of Ad vectors possessing augmented tumor cell transduction properties.246, 247, 248, 249
A substantial fraction of OS tumors contain cells expressing low levels of CAR and have thus demonstrated resistance to conventional Ad vectors.250, 251 Witlox et al252 observed that primary OS tumor cells express high levels of integrins avb3 and avb5, and generated an adenoviral vector containing an Arg-Gly-Asp (RGD-4C) integrin-targeting motif (AdCMVlucRGD) in the fiber knob. They showed that this retargeted Ad vector transduces OS cells about 100-fold more efficiently than unmodified Ad5. Testing a panel of Ad vectors recently generated in our lab, we found that replacement of the human knob domain of the Ad5 fiber with knob domains of canine adenoviruses CAV1 and CAV2 resulted in an augmentation of transduction efficiency of human OS cells (V Ternovoi, unpublished data, 2005).253, 254 Particularly interesting are results with a fiber mosaic Ad5 vector encoding two fibers: the σ1 and the wt Ad5 fiber.255 This mosaic transduces some primary OS cell lines even more efficiently than AdCMVlucRGD (V Ternovoi, unpublished data, 2005).
Recently, selective targeting of cognate antigen has been achieved via genetic incorporation of a cytosol stable single chain fragments of antibody (scFv), known as an intrabody, into a deknobbed Ad fiber.256 In this regard, a scFv derived from the monoclonal antibody 8H9 (MAb 8H9) could be an excellent candidate for specific targeting of OS cells. The antigen epitope recognized by the 8H9 mouse antibody is a putative cell surface glycoprotein expressed on cell membranes in a broad spectrum of tumors of neuroectodermal, mesenchymal and epithelial origin, with restricted distribution in normal tissues.257 Importantly, 28 of 29 human OS have tested positive with 8H9 by immunohistochemistry. Recombinant immunotoxin containing the 8H9 scFv selectively killed cells that react with the MAb 8H9 and produced regression of human cancers growing in severe combined immunodeficient mice that express the 8H9 antigen.258 Particularly important, the dose that caused significant regression in mice was well tolerated in monkeys.
Animal models
The immune system may play an important role in the realization of p53's overall antitumor potential. As has been discussed above, p53 overexpression in the context of viral vectors can induce a powerful antitumor response. This response can even result in the rejection of well-established tumors, but only in the context of immunomodulation strategies aimed at overcoming the tolerogenic activity of the tumor microenvironment. These results are encouraging, but they have been achieved using animals with experimentally established tumors. Spontaneous tumors present a significantly more complex challenge for induction of an efficient immune response.259 They are engaged in the process of successfully fighting against host defense systems and are comprised of a population of heterogenous tumor cells and recruited host stromal cells. Stromal cells, including fibroblasts, inflammatory cells, and vascular cells, generate a microenvironment that actively fosters tumor growth and prevents an immune response against these same tumor cells.260, 261, 262, 263, 264, 265, 266 Together, the heterogenous tumor cell populations, shaped by the immune system in the process of multistep progression,267, 268 and considering the tumor microenvironment which actively induces the status of immune tolerance,269 allow exceptional establishment of spontaneous tumors, which experimentally established tumors cannot reproduce. Thus, (1) generation of an efficient antitumor immune response will be much harder to achieve with naturally occurring tumors and (2) to get clinically relevant information regarding therapeutic efficacy of p53 tumor suppressor gene therapy, its preclinical evaluation should be carried out using immunocompetent animals with spontaneously developed OS.
An immunocompetent animal model could also be helpful in finding a solution for the problem that results from high immunogenicity of Ad vectors. Adenoviruses induce a potent immune response which significantly reduces efficiency of Ad-based vectors.270, 271 Ablation of immune response have been shown to facilitate oncolytic virus intratumoral propagation.249, 272, 273 Consequently, immunosuppression would be beneficial for CRAd intratumoral spreading and expression of p53 in this context should result in a significantly more potent tumor cell killing via apoptogenic activity of p53. In contradistinction, a realization exists that the tumor suppressor effect of p53 relies on the induction of an antitumor immune response. Thus immunosuppression should diminish antitumor potential. One of the strategies to solve this problem might be induction of temporal immunoablation in the first phase of treatment followed by immunostimulation to achieve an active Th1 immune response against tumor antigens. In this scenario, the Ad vector will get opportunity to achieve tumor penetration and spread, kill a significant fraction of tumor cells, and subsequent administration of immunopotentiating agents with Th1-polarizing activity would augment an antitumor response.
Canine OS bears striking resemblance to human OS,274, 275 and in our hands and others appears to be a useful animal model for testing gene therapy approaches because dogs share many physiological features with humans.276 Greatly accelerated kinetics of tumor growth and progression in dogs relative to humans creates the opportunity to assess treatment outcomes in a reasonable time frame. To study host–vector interactions and to develop strategies facilitating Ad vector intratumoral replication in the face of an active immune system, we generated a CRAd agent based on the canine adenovirus type 2 genome.277 Interestingly, we have also found that human adenovirus type 5 can productively infect canine tumor cells of different tissue origins. Furthermore, we provided evidence of Ad5 lateral spreading in primary canine OS cells.278 These findings raise the probability of exploiting canine OS as a model for preclinical analysis of candidate Ad5 based vectors designed for human OS gene therapy.
Concluding remarks and future directions
OS is often resistant to conventional multimodal therapy for malignant disease. Expression profiling of chemoresistant OS has results in partial unmasking of its resistance signature. The gene expression profile and other properties of resistance to therapy closely resemble properties of cancer stem cells. Stem cell populations have been identified in a range of hematopoietic and solid tumors, and the capacity of these cells for unlimited self-renewal and intrinsic resistance to cell death is now recognized as important mechanisms of tumor resistance to current therapy.
Adenovirus-mediated p53 tumor suppressor gene therapy is a newer treatment modality with high antitumor potential. In fact, this is a combined modality which employes several antitumor mechanisms: p53 ‘intrinsic’ and ‘extrinsic’ tumor suppressor mechanisms and cytolytic activity of replication competent Ad. However, analysis of the peer-reviewed literature revealed that wt p53 and currently used Ad vectors may not be efficient tools for OS treatment because OS cells generally overexpress inhibitors of wt p53 and express low levels of the CAR receptor. Recent advances in understanding the molecular mechanisms of p53's proapoptotic action suggest that cointroduction of a modified p53 gene resistant to inhibitors (Mdm2 and adenoviral protein E1B-55K) with the PTEN gene into tumor cells should result in efficient apoptosis induction in these tumor cells with a high apoptotic threshold. The clinical relevance of this therapeutic approach will be higher if infection of a significant fraction of the tumor cells can be achieved. Temporal immunoablation to facilitate CRAd agent tumor targeting and intratumoral spreading with subsequent immunopotentiation to overcome the immunosuppressive activity of the tumor microenvironment, while boosting the immune response against tumor antigens, presents one possible solution to this problem.
The complexity of factors influencing the final outcome of this new modality and its reliance on immune mechanisms requires employment of an adequate model, and canine OS may represent the ideal preclinical model for validation of adenovirus-mediated p53 tumor suppressor gene therapy. Further insights into the mechanisms of tumor progression, tumor–stroma interactions and tumor–immune system relationships, along with p53's biology, host–vector and virus-cell signal transduction interplay will result in more rationally designed therapeutic strategies with increased clinical benefits for OS patients.
References
Dorfman HD, Czerniak B . Bone cancers. Cancer 1995;75:203–210.
Link MPEF . Osteosarcoma. In: Pizzo PAPD (ed). Principles and Practice of Pediatric Oncology. Lippincott-Raven: Philadelphia, PA, 1997, pp 889–920.
Rosen GF, Forcher CA, Mankin HJ, et al. Neoplasm of bone and soft tissue. In: Bast RC, Holland JF, Frei E et al (eds). Cancer Medicine. B.C. Decker Inc: Hamilton, Canada, 2000, pp 1870–1902.
Meyers PA, Gorlick R . Osteosarcoma. Pediatr Clin North Am 1997;44:973–989.
Ferguson WS, Goorin AM . Current treatment of osteosarcoma. Cancer Invest 2001;19:292–315.
Bacci G, Forni C, Ferrari S, et al. Neoadjuvant chemotherapy for osteosarcoma of the extremity: intensification of preoperative treatment does not increase the rate of good histologic response to the primary tumor or improve the final outcome. J Pediatr Hematol Oncol 2003;25:845–853.
Gorlick R, Meyers PA . Osteosarcoma necrosis following chemotherapy: innate biology versus treatment-specific. J Pediatr Hematol Oncol 2003;25:840–841.
Smeland S, Muller C, Alvegard TA, et al. Scandinavian Sarcoma Group Osteosarcoma Study SSG VIII: prognostic factors for outcome and the role of replacement salvage chemotherapy for poor histological responders. Eur J Cancer 2003;39:488–494.
Sherr CJ . Principles of tumor suppression. Cell 2004;116:235–246.
Roemer K, Friedmann T . Mechanisms of action of the p53 tumor suppressor and prospects for cancer gene therapy by reconstitution of p53 function. Ann NY Acad Sci 1994;716:265–280; discussion 280–262.
McCormick F . Cancer gene therapy: fringe or cutting edge? Nat Rev Cancer 2001;1:130–141.
Mudry RE, Fortney JE, York T, et al. Stromal cells regulate survival of B-lineage leukemic cells during chemotherapy. Blood 2000;96:1926–1932.
Waldman T, Zhang Y, Dillehay L, et al. Cell-cycle arrest versus cell death in cancer therapy. Nat Med 1997;3:1034–1036.
Wang E . Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res 1995;55:2284–2292.
Van Meir EG, Polverini PJ, Chazin VR, et al. Release of an inhibitor of angiogenesis upon induction of wild type p53 expression in glioblastoma cells. Nat Genet 1994;8:171–176.
Takaoka A, Hayakawa S, Yanai H, et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003;424:516–523.
Nielsen LL, Maneval DC . P53 tumor suppressor gene therapy for cancer. Cancer Gene Ther 1998;5:52–63.
McNeish IA, Bell SJ, Lemoine NR . Gene therapy progress and prospects: cancer gene therapy using tumour suppressor genes. Gene Therapy 2004;11:497–503.
Peng Z . Current status of gendicine in China: recombinant human Ad-p53 agent for treatment of cancers. Hum Gene Ther 2005;16:1016–1027.
Roth JA . Adenovirus p53 gene therapy. Expert Opin Biol Ther 2006;6:55–61.
Zeimet AG, Marth C . Why did p53 gene therapy fail in ovarian cancer? Lancet Oncol 2003;4:415–422.
Atencio IA, Grace M, Bordens R, et al. Biological activities of a recombinant adenovirus p53 (SCH 58500) administered by hepatic arterial infusion in a Phase 1 colorectal cancer trial. Cancer Gene Ther 2005;13:169–181.
Lang FF, Bruner JM, Fuller GN, et al. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol 2003;21:2508–2518.
Pisters LL, Pettaway CA, Troncoso P, et al. Evidence that transfer of functional p53 protein results in increased apoptosis in prostate cancer. Clin Cancer Res 2004;10:2587–2593.
Sionov RV, Haupt Y . The cellular response to p53: the decision between life and death. Oncogene 1999;18:6145–6157.
Oren M . Decision making by p53: life, death and cancer. Cell Death Differ 2003;10:431–442.
Vogelstein B, Kinzler KW . p53 function and dysfunction. Cell 1992;70:523–526.
Donehower LA, Bradley A . The tumor suppressor p53. Biochim Biophys Acta 1993;1155:181–205.
Levine AJ . p53, the cellular gatekeeper for growth and division. Cell 1997;88:323–331.
Cadwell C, Zambetti GP . The effects of wild-type p53 tumor suppressor activity and mutant p53 gain-of-function on cell growth. Gene 2001;277:15–30.
Prives C, Hall PA . The p53 pathway. J Pathol 1999;187:112–126.
Lin J, Chen J, Elenbaas B, et al. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev 1994;8:1235–1246.
Haupt Y, Maya R, Kazaz A, et al. Mdm2 promotes the rapid degradation of p53. Nature 1997;387:296–299.
Kubbutat MH, Jones SN, Vousden KH . Regulation of p53 stability by Mdm2. Nature 1997;387:299–303.
Candau R, Scolnick DM, Darpino P, et al. Two tandem and independent sub-activation domains in the amino terminus of p53 require the adaptor complex for activity. Oncogene 1997;15:807–816.
Chen X, Ko LJ, Jayaraman L, et al. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev 1996;10:2438–2451.
Sakamuro D, Sabbatini P, White E, et al. The polyproline region of p53 is required to activate apoptosis but not growth arrest. Oncogene 1997;15:887–898.
Venot C, Maratrat M, Dureuil C, et al. The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J 1998;17:4668–4679.
Zhu J, Jiang J, Zhou W, et al. Differential regulation of cellular target genes by p53 devoid of the PXXP motifs with impaired apoptotic activity. Oncogene 1999;18:2149–2155.
Giaccia AJ, Kastan MB . The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev 1998;12:2973–2983.
Harris SL, Levine AJ . The p53 pathway: positive and negative feedback loops. Oncogene 2005;24:2899–2908.
Ko LJ, Prives C . p53: puzzle and paradigm. Genes Dev 1996;10:1054–1072.
de Vries A, Flores ER, Miranda B, et al. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc Natl Acad Sci USA 2002;99:2948–2953.
Sigal A, Rotter V . Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res 2000;60:6788–6793.
Blandino G, Levine AJ, Oren M . Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene 1999;18:477–485.
Woods DB, Vousden KH . Regulation of p53 function. Exp Cell Res 2001;264:56–66.
Momand J, Wu HH, Dasgupta G . Mdm2—master regulator of the p53 tumor suppressor protein. Gene 2000;242:15–29.
Chen J, Lin J, Levine AJ . Regulation of transcription functions of the p53 tumor suppressor by the mdm-2 oncogene. Mol Med 1995;1:142–152.
Momand J, Zambetti GP, Olson DC, et al. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992;69:1237–1245.
Barak Y, Juven T, Haffner R, et al. mdm2 expression is induced by wild type p53 activity. EMBO J 1993;12:461–468.
Wu X, Bayle JH, Olson D, et al. The p53-mdm-2 autoregulatory feedback loop. Genes Dev 1993;7:1126–1132.
Jones SN, Hancock AR, Vogel H, et al. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc Natl Acad Sci USA 1998;95:15608–15612.
Sherr CJ, Weber JD . The ARF/p53 pathway. Curr Opin Genet Dev 2000;10:94–99.
Lohrum MA, Ludwig RL, Kubbutat MH, et al. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 2003;3:577–587.
Okamoto K, Li H, Jensen MR, et al. Cyclin G recruits PP2A to dephosphorylate Mdm2. Mol Cell 2002;9:761–771.
Higashitsuji H, Itoh K, Sakurai T, et al. The oncoprotein gankyrin binds to Mdm2/HDM2, enhancing ubiquitylation and degradation of p53. Cancer Cell 2005;8:75–87.
Sui G, Affar el B, Shi Y, et al. Yin Yang 1 is a negative regulator of p53. Cell 2004;117:859–872.
Wang C, Ivanov A, Chen L, et al. Mdm2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J 2005;24:3279–3290.
Bergamaschi D, Gasco M, Hiller L, et al. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell 2003;3:387–402.
El-Deiry WS . The role of p53 in chemosensitivity and radiosensitivity. Oncogene 2003;22:7486–7495.
Fridman JS, Lowe SW . Control of apoptosis by p53. Oncogene 2003;22:9030–9040.
Slee EA, O'Connor DJ, Lu X . To die or not to die: how does p53 decide? Oncogene 2004;23:2809–2818.
Vousden KH, Lu X . Live or let die: the cell's response to p53. Nat Rev Cancer 2002;2:594–604.
Miyashita T, Krajewski S, Krajewska M, et al. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 1994;9:1799–1805.
Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001;292:727–730.
Nakano K, Vousden KH . PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 2001;7:683–694.
Yu J, Wang Z, Kinzler KW, et al. PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc Natl Acad Sci USA 2003;100:1931–1936.
Oda E, Ohki R, Murasawa H, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000;288:1053–1058.
Sax JK, Fei P, Murphy ME, et al. BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol 2002;4:842–849.
Moroni MC, Hickman ES, Lazzerini Denchi E, et al. Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol 2001;3:552–558.
MacLachlan TK, Takimoto R, El-Deiry WS . BRCA1 directs a selective p53-dependent transcriptional response towards growth arrest and DNA repair targets. Mol Cell Biol 2002;22:4280–4292.
Takimoto R, El-Deiry WS . Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 2000;19:1735–1743.
Tanikawa C, Matsuda K, Fukuda S, et al. p53RDL1 regulates p53-dependent apoptosis. Nat Cell Biol 2003;5:216–223.
el-Deiry WS . Regulation of p53 downstream genes. Semin Cancer Biol 1998;8:345–357.
Stambolic V, MacPherson D, Sas D, et al. Regulation of PTEN transcription by p53. Mol Cell 2001;8:317–325.
Erster S, Moll UM . Stress-induced p53 runs a transcription-independent death program. Biochem Biophys Res Commun 2005;331:843–850.
Moll UM, Wolff S, Speidel D, et al. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol 2005;17:631–636.
Yee KS, Vousden KH . Complicating the complexity of p53. Carcinogenesis 2005;26:1317–1322.
Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003;11:577–590.
Chipuk JE, Kuwana T, Bouchier-Hayes L, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004;303:1010–1014.
Chipuk JE, Bouchier-Hayes L, Kuwana T, et al. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 2005;309:1732–1735.
Dumont P, Leu JI, Della Pietra III AC, et al. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet 2003;33:357–365.
Mack DH, Vartikar J, Pipas JM, et al. Specific repression of TATA-mediated but not initiator-mediated transcription by wild-type p53. Nature 1993;363:281–283.
Zhang CC, Yang JM, Bash-Babula J, et al. DNA damage increases sensitivity to vinca alkaloids and decreases sensitivity to taxanes through p53-dependent repression of microtubule-associated protein 4. Cancer Res 1999;59:3663–3670.
Kho PS, Wang Z, Zhuang L, et al. p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J Biol Chem 2004;279:21183–21192.
Shikama N, Lee CW, France S, et al. A novel cofactor for p300 that regulates the p53 response. Mol Cell 1999;4:365–376.
Samuels-Lev Y, O'Connor DJ, Bergamaschi D, et al. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell 2001;8:781–794.
Flores ER, Tsai KY, Crowley D, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002;416:560–564.
Dyson N . The regulation of E2F by pRB-family proteins. Genes Dev 1998;12:2245–2262.
Helin K . Regulation of cell proliferation by the E2F transcription factors. Curr Opin Genet Dev 1998;8:28–35.
Stevaux O, Dyson NJ . A revised picture of the E2F transcriptional network and RB function. Curr Opin Cell Biol 2002;14:684–691.
Trimarchi JM, Lees JA . Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol 2002;3:11–20.
Dimova DK, Dyson NJ . The E2F transcriptional network: old acquaintances with new faces. Oncogene 2005;24:2810–2826.
Ginsberg D . E2F1 pathways to apoptosis. FEBS Lett 2002;529:122–125.
Berkovich E, Ginsberg D . ATM is a target for positive regulation by E2F-1. Oncogene 2003;22:161–167.
Rogoff HA, Pickering MT, Frame FM, et al. Apoptosis associated with deregulated E2F activity is dependent on E2F1 and Atm/Nbs1/Chk2. Mol Cell Biol 2004;24:2968–2977.
Fogal V, Kartasheva NN, Trigiante G, et al. ASPP1 and ASPP2 are new transcriptional targets of E2F. Cell Death Differ 2005;12:369–376.
Hershko T, Chaussepied M, Oren M, et al. Novel link between E2F and p53: proapoptotic cofactors of p53 are transcriptionally upregulated by E2F. Cell Death Differ 2005;12:377–383.
Hershko T, Ginsberg D . Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J Biol Chem 2004;279:8627–8634.
Fortin A, MacLaurin JG, Arbour N, et al. The proapoptotic gene SIVA is a direct transcriptional target for the tumor suppressors p53 and E2F1. J Biol Chem 2004;279:28706–28714.
Zaika A, Irwin M, Sansome C, et al. Oncogenes induce and activate endogenous p73 protein. J Biol Chem 2001;276:11310–11316.
La Sala D, Macaluso M, Trimarchi C, et al. Triggering of p73-dependent apoptosis in osteosarcoma is under the control of E2Fs-pRb2/p130 complexes. Oncogene 2003;22:3518–3529.
Sowers R, Toguchida J, Qin J, et al. mRNA expression levels of E2F transcription factors correlate with dihydrofolate reductase, reduced folate carrier, and thymidylate synthase mRNA expression in osteosarcoma. Mol Cancer Ther 2003;2:535–541.
Di Cristofano A, Pandolfi PP . The multiple roles of PTEN in tumor suppression. Cell 2000;100:387–390.
Cantley LC, Neel BG . New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA 1999;96:4240–4245.
Hamada K, Sasaki T, Koni PA, et al. The PTEN/PI3K pathway governs normal vascular development and tumor angiogenesis. Genes Dev 2005;19:2054–2065.
Mayo LD, Seo YR, Jackson MW, et al. Phosphorylation of human p53 at serine 46 determines promoter selection and whether apoptosis is attenuated or amplified. J Biol Chem 2005;280:25953–25959.
Gottlieb TM, Leal JF, Seger R, et al. Cross-talk between Akt, p53 and Mdm2: possible implications for the regulation of apoptosis. Oncogene 2002;21:1299–1303.
Chang CJ, Freeman DJ, Wu H . PTEN regulates Mdm2 expression through the P1 promoter. J Biol Chem 2004;279:29841–29848.
Freeman DJ, Li AG, Wei G, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell 2003;3:117–130.
Zhou M, Gu L, Findley HW, et al. PTEN reverses Mdm2-mediated chemotherapy resistance by interacting with p53 in acute lymphoblastic leukemia cells. Cancer Res 2003;63:6357–6362.
Ali IU, Schriml LM, Dean M . Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst 1999;91:1922–1932.
Yamaguchi T, Toguchida J, Yamamuro T, et al. Allelotype analysis in osteosarcomas: frequent allele loss on 3q, 13q, 17p, and 18q. Cancer Res 1992;52:2419–2423.
Coates PJ, Lorimore SA, Wright EG . Cell and tissue responses to genotoxic stress. J Pathol 2005;205:221–235.
Jacks T, Weinberg RA . Taking the study of cancer cell survival to a new dimension. Cell 2002;111:923–925.
Datta SR, Brunet A, Greenberg ME . Cellular survival: a play in three Akts. Genes Dev 1999;13:2905–2927.
Vivanco I, Sawyers CL . The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2002;2:489–501.
Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998;282:1318–1321.
Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999;96:857–868.
Brunet A, Datta SR, Greenberg ME . Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 2001;11:297–305.
Boudreau N, Werb Z, Bissell MJ . Suppression of apoptosis by basement membrane requires three-dimensional tissue organization and withdrawal from the cell cycle. Proc Natl Acad Sci USA 1996;93:3509–3513.
Bissell MJ, Kenny PA, Radisky DC . Microenvironmental regulators of tissue structure and function also regulate tumor induction and progression: the role of extracellular matrix and its degrading enzymes. Cold Spring Harb Symp Quant Biol 2005;70:1–14.
Zahir N, Weaver VM . Death in the third dimension: apoptosis regulation and tissue architecture. Curr Opin Genet Dev 2004;14:71–80.
Wang F, Hansen RK, Radisky D, et al. Phenotypic reversion or death of cancer cells by altering signaling pathways in three-dimensional contexts. J Natl Cancer Inst 2002;94:1494–1503.
Dameron KM, Volpert OV, Tainsky MA, et al. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science 1994;265:1582–1584.
Mashimo T, Watabe M, Hirota S, et al. The expression of the KAI1 gene, a tumor metastasis suppressor, is directly activated by p53. Proc Natl Acad Sci USA 1998;95:11307–11311.
Nishimori H, Shiratsuchi T, Urano T, et al. A novel brain-specific p53-target gene, BAI1, containing thrombospondin type 1 repeats inhibits experimental angiogenesis. Oncogene 1997;15:2145–2150.
Zou Z, Gao C, Nagaich AK, et al. p53 regulates the expression of the tumor suppressor gene maspin. J Biol Chem 2000;275:6051–6054.
Bouvet M, Ellis LM, Nishizaki M, et al. Adenovirus-mediated wild-type p53 gene transfer down-regulates vascular endothelial growth factor expression and inhibits angiogenesis in human colon cancer. Cancer Res 1998;58:2288–2292.
Harada H, Nakagawa K, Saito M, et al. Introduction of wild-type p53 enhances thrombospondin-1 expression in human glioma cells. Cancer Lett 2003;191:109–119.
Nishizaki M, Fujiwara T, Tanida T, et al. Recombinant adenovirus expressing wild-type p53 is antiangiogenic: a proposed mechanism for bystander effect. Clin Cancer Res 1999;5:1015–1023.
Riccioni T, Cirielli C, Wang X, et al. Adenovirus-mediated wild-type p53 overexpression inhibits endothelial cell differentiation in vitro and angiogenesis in vivo. Gene Therapy 1998;5:747–754.
Zhang L, Yu D, Hu M, et al. Wild-type p53 suppresses angiogenesis in human leiomyosarcoma and synovial sarcoma by transcriptional suppression of vascular endothelial growth factor expression. Cancer Res 2000;60:3655–3661.
Gautam A, Densmore CL, Melton S, et al. Aerosol delivery of PEI-p53 complexes inhibits B16-F10 lung metastases through regulation of angiogenesis. Cancer Gene Ther 2002;9:28–36.
Mecchia M, Matarrese P, Malorni W, et al. Type I consensus interferon (CIFN) gene transfer into human melanoma cells up-regulates p53 and enhances cisplatin-induced apoptosis: implications for new therapeutic strategies with IFN-alpha. Gene Therapy 2000;7:167–179.
Mori T, Anazawa Y, Iiizumi M, et al. Identification of the interferon regulatory factor 5 gene (IRF-5) as a direct target for p53. Oncogene 2002;21:2914–2918.
Schoenemeyer A, Barnes BJ, Mancl ME, et al. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J Biol Chem 2005;280:17005–17012.
Clemens MJ . Interferons and apoptosis. J Interferon Cytokine Res 2003;23:277–292.
Pestka S, Krause CD, Walter MR . Interferons, interferon-like cytokines, and their receptors. Immunol Rev 2004;202:8–32.
Carroll JL, Nielsen LL, Pruett SB, et al. The role of natural killer cells in adenovirus-mediated p53 gene therapy. Mol Cancer Ther 2001;1:49–60.
Fodor I, Timiryasova T, Denes B, et al. Vaccinia virus mediated p53 gene therapy for bladder cancer in an orthotopic murine model. J Urol 2005;173:604–609.
Li Z, Rakkar A, Katayose Y, et al. Efficacy of multiple administrations of a recombinant adenovirus expressing wild-type p53 in an immune-competent mouse tumor model. Gene Therapy 1998;5:605–613.
Roth J, Dittmer D, Rea D, et al. p53 as a target for cancer vaccines: recombinant canarypox virus vectors expressing p53 protect mice against lethal tumor cell challenge. Proc Natl Acad Sci USA 1996;93:4781–4786.
Blaszczyk-Thurin M, Ertl IO, Ertl HC . An experimental vaccine expressing wild-type p53 induces protective immunity against glioblastoma cells with high levels of endogenous p53. Scand J Immunol 2002;56:361–375.
Daftarian P, Song GY, Ali S, et al. Two distinct pathways of immuno-modulation improve potency of p53 immunization in rejecting established tumors. Cancer Res 2004;64:5407–5414.
Vierboom MP, Nijman HW, Offringa R, et al. Tumor eradication by wild-type p53-specific cytotoxic T lymphocytes. J Exp Med 1997;186:695–704.
Rosenberg SA, Yang JC, Restifo NP . Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004;10:909–915.
Almog N, Rotter V . Involvement of p53 in cell differentiation and development. Biochim Biophys Acta 1997;1333:F1–F27.
Attardi LD, Donehower LA . Probing p53 biological functions through the use of genetically engineered mouse models. Mutat Res 2005;576:4–21.
Choi J, Donehower LA . p53 in embryonic development: maintaining a fine balance. Cell Mol Life Sci 1999;55:38–47.
Murray-Zmijewski F, Lane DP, Bourdon JC . p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ 2006 [Epub ahead of print].
Lin T, Chao C, Saito S, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol 2005;7:165–171.
Aladjem MI, Spike BT, Rodewald LW, et al. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr Biol 1998;8:145–155.
Rambhatla L, Ram-Mohan S, Cheng JJ, et al. Immortal DNA strand cosegregation requires p53/IMPDH-dependent asymmetric self-renewal associated with adult stem cells. Cancer Res 2005;65:3155–3161.
Gil-Perotin S, Marin-Husstege M, Li J, et al. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci 2006;26:1107–1116.
Meletis K, Wirta V, Hede SM, et al. p53 suppresses the self-renewal of adult neural stem cells. Development 2006;133:363–369.
Ohyama K, Chung CH, Chen E, et al. p53 influences mice skeletal development. J Craniofac Genet Dev Biol 1997;17:161–171.
Gorlick R, Anderson P, Andrulis I, et al. Biology of childhood osteogenic sarcoma and potential targets for therapeutic development: meeting summary. Clin Cancer Res 2003;9:5442–5453.
Ragland BD, Bell WC, Lopez RR, et al. Cytogenetics and molecular biology of osteosarcoma. Lab Invest 2002;82:365–373.
Chandar N, Campbell P, Novak J, et al. Dependence of induction of osteocalcin gene expression on the presence of wild-type p53 in a murine osteosarcoma cell line. Mol Carcinog 1993;8:299–305.
Tataria M, Quarto N, Longaker MT, et al. Absence of the p53 tumor suppressor gene promotes osteogenesis in mesenchymal stem cells. J Pediatr Surg 2006;41:624–632; discussion 624–632.
Lengner CJ, Steinman HA, Gagnon J, et al. Osteoblast differentiation and skeletal development are regulated by Mdm2-p53 signaling. J Cell Biol 2006;172:909–921.
Nakashima K, Zhou X, Kunkel G, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002;108:17–29.
Wang X, Kua HY, Hu Y, et al. p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. J Cell Biol 2006;172:115–125.
Stein GS, Lian JB, van Wijnen AJ, et al. Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene 2004;23:4315–4329.
Blyth K, Cameron ER, Neil JC . The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer 2005;5:376–387.
Ducy P, Zhang R, Geoffroy V, et al. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 1997;89:747–754.
Komori T . Runx2, a multifunctional transcription factor in skeletal development. J Cell Biochem 2002;87:1–8.
Takeda S, Bonnamy JP, Owen MJ, et al. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev 2001;15:467–481.
Pratap J, Galindo M, Zaidi SK, et al. Cell growth regulatory role of Runx2 during proliferative expansion of preosteoblasts. Cancer Res 2003;63:5357–5362.
Galindo M, Pratap J, Young DW, et al. The bone-specific expression of Runx2 oscillates during the cell cycle to support a G1-related antiproliferative function in osteoblasts. J Biol Chem 2005;280:20274–20285.
Komori T . Regulation of skeletal development by the Runx family of transcription factors. J Cell Biochem 2005;95:445–453.
Thomas DM, Johnson SA, Sims NA, et al. Terminal osteoblast differentiation, mediated by runx2 and p27KIP1, is disrupted in osteosarcoma. J Cell Biol 2004;167:925–934.
Cordenonsi M, Dupont S, Maretto S, et al. Links between tumor suppressors: p53 is required for TGF-beta gene responses by cooperating with Smads. Cell 2003;113:301–314.
Fero ML, Randel E, Gurley KE, et al. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 1998;396:177–180.
Philipp-Staheli J, Kim KH, Liggitt D, et al. Distinct roles for p53, p27Kip1, and p21Cip1 during tumor development. Oncogene 2004;23:905–913.
Bellido T, Ali AA, Plotkin LI, et al. Proteasomal degradation of Runx2 shortens parathyroid hormone-induced anti-apoptotic signaling in osteoblasts. A putative explanation for why intermittent administration is needed for bone anabolism. J Biol Chem 2003;278:50259–50272.
Blyth K, Vaillant F, Hanlon L, et al. Runx2 and MYC collaborate in lymphoma development by suppressing apoptotic and growth arrest pathways in vivo. Cancer Res 2006;66:2195–2201.
Chandar N, Donehower L, Lanciloti N . Reduction in p53 gene dosage diminishes differentiation capacity of osteoblasts. Anticancer Res 2000;20:2553–2559.
Chandar N, Swindle J, Szajkovics A, et al. Relationship of bone morphogenetic protein expression during osteoblast differentiation to wild type p53. J Orthop Res 2005;23:1345–1353.
Chandar N, Saluja R, Lamar PC, et al. P53 and beta-catenin activity during estrogen treatment of osteoblasts. Cancer Cell Int 2005;5:24.
Boland GM, Perkins G, Hall DJ, et al. Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. J Cell Biochem 2004;93:1210–1230.
Day TF, Guo X, Garrett-Beal L, et al. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell 2005;8:739–750.
Kahler RA, Westendorf JJ . Lymphoid enhancer factor-1 and beta-catenin inhibit Runx2-dependent transcriptional activation of the osteocalcin promoter. J Biol Chem 2003;278:11937–11944.
Vaes BL, Dechering KJ, van Someren EP, et al. Microarray analysis reveals expression regulation of Wnt antagonists in differentiating osteoblasts. Bone 2005;36:803–811.
Westendorf JJ, Kahler RA, Schroeder TM . Wnt signaling in osteoblasts and bone diseases. Gene 2004;341:19–39.
Logan CY, Nusse R . The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004;20:781–810.
Haydon RC, Deyrup A, Ishikawa A, et al. Cytoplasmic and/or nuclear accumulation of the beta-catenin protein is a frequent event in human osteosarcoma. Int J Cancer 2002;102:338–342.
Overholtzer M, Rao PH, Favis R, et al. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc Natl Acad Sci USA 2003;100:11547–11552.
Olive KP, Tuveson DA, Ruhe ZC, et al. Mutant p53 gain of function in two mouse models of Li–Fraumeni syndrome. Cell 2004;119:847–860.
Lang GA, Iwakuma T, Suh YA, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004;119:861–872.
Man TK, Chintagumpala M, Visvanathan J, et al. Expression profiles of osteosarcoma that can predict response to chemotherapy. Cancer Res 2005;65:8142–8150.
Mintz MB, Sowers R, Brown KM, et al. An expression signature classifies chemotherapy-resistant pediatric osteosarcoma. Cancer Res 2005;65:1748–1754.
Ochi K, Daigo Y, Katagiri T, et al. Prediction of response to neoadjuvant chemotherapy for osteosarcoma by gene-expression profiles. Int J Oncol 2004;24:647–655.
Bialek P, Kern B, Yang X, et al. A twist code determines the onset of osteoblast differentiation. Dev Cell 2004;6:423–435.
Yousfi M, Lasmoles F, Marie PJ . TWIST inactivation reduces CBFA1/RUNX2 expression and DNA binding to the osteocalcin promoter in osteoblasts. Biochem Biophys Res Commun 2002;297:641–644.
Maestro R, Dei Tos AP, Hamamori Y, et al. Twist is a potential oncogene that inhibits apoptosis. Genes Dev 1999;13:2207–2217.
Valsesia-Wittmann S, Magdeleine M, Dupasquier S, et al. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell 2004;6:625–630.
Wang X, Ling MT, Guan XY, et al. Identification of a novel function of TWIST, a bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogene 2004;23:474–482.
Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004;117:927–939.
Lee MS, Lowe GN, Strong DD, et al. TWIST, a basic helix–loop–helix transcription factor, can regulate the human osteogenic lineage. J Cell Biochem 1999;75:566–577.
Dean M, Fojo T, Bates S . Tumour stem cells and drug resistance. Nat Rev Cancer 2005;5:275–284.
Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature 2001;414:105–111.
Chen PL, Chen YM, Bookstein R, et al. Genetic mechanisms of tumor suppression by the human p53 gene. Science 1990;250:1576–1580.
Marcellus RC, Teodoro JG, Charbonneau R, et al. Expression of p53 in Saos-2 osteosarcoma cells induces apoptosis which can be inhibited by Bcl-2 or the adenovirus E1B-55 kDa protein. Cell Growth Differ 1996;7:1643–1650.
Tsuchiya H, Mori Y, Ueda Y, et al. Sensitization and caffeine potentiation of cisplatin cytotoxicity resulting from introduction of wild-type p53 gene in human osteosarcoma. Anticancer Res 2000;20:235–242.
Wang J, Bucana CD, Roth JA, et al. Apoptosis induced in human osteosarcoma cells is one of the mechanisms for the cytocidal effect of Ad5CMV-p53. Cancer Gene Ther 1995;2:9–17.
Wills KN, Maneval DC, Menzel P, et al. Development and characterization of recombinant adenoviruses encoding human p53 for gene therapy of cancer. Hum Gene Ther 1994;5:1079–1088.
Densmore CL, Kleinerman ES, Gautam A, et al. Growth suppression of established human osteosarcoma lung metastases in mice by aerosol gene therapy with PEI-p53 complexes. Cancer Gene Ther 2001;8:619–627.
Song SU, Boyce FM . Combination treatment for osteosarcoma with baculoviral vector mediated gene therapy (p53) and chemotherapy (adriamycin). Exp Mol Med 2001;33:46–53.
Ganjavi H, Gee M, Narendran A, et al. Adenovirus-mediated p53 gene therapy in osteosarcoma cell lines: sensitization to cisplatin and doxorubicin. Cancer Gene Ther 2005;13:415–419.
Hellwinkel OJ, Muller J, Pollmann A, et al. Osteosarcoma cell lines display variable individual reactions on wildtype p53 and Rb tumour-suppressor transgenes. J Gene Med 2005;7:407–419.
Oliner JD, Kinzler KW, Meltzer PS, et al. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992;358:80–83.
Kawashima H, Ogose A, Yoshizawa T, et al. Expression of the coxsackievirus and adenovirus receptor in musculoskeletal tumors and mesenchymal tissues: efficacy of adenoviral gene therapy for osteosarcoma. Cancer Sci 2003;94:70–75.
van Beusechem VW, van den Doel PB, Gerritsen WR . Conditionally replicative adenovirus expressing degradation-resistant p53 for enhanced oncolysis of human cancer cells overexpressing murine double minute 2. Mol Cancer Ther 2005;4:1013–1018.
Witlox MA, Van Beusechem VW, Grill J, et al. Epidermal growth factor receptor targeting enhances adenoviral vector based suicide gene therapy of osteosarcoma. J Gene Med 2002;4:510–516.
Conseiller E, Debussche L, Landais D, et al. CTS1: a p53-derived chimeric tumor suppressor gene with enhanced in vitro apoptotic properties. J Clin Invest 1998;101:120–127.
Bougeret C, Virone-Oddos A, Adeline E, et al. Cancer gene therapy mediated by CTS1, a p53 derivative: advantage over wild-type p53 in growth inhibition of human tumors overexpressing Mdm2. Cancer Gene Ther 2000;7:789–798.
Tang HJ, Qian D, Sondak VK, et al. A modified p53 enhances apoptosis in sarcoma cell lines mediated by doxorubicin. Br J Cancer 2004;90:1285–1292.
Alemany R, Balague C, Curiel DT . Replicative adenoviruses for cancer therapy. Nat Biotechnol 2000;18:723–727.
Hermiston TW, Kuhn I . Armed therapeutic viruses: strategies and challenges to arming oncolytic viruses with therapeutic genes. Cancer Gene Ther 2002;9:1022–1035.
Sarnow P, Ho YS, Williams J, et al. Adenovirus E1b-58kd tumor antigen and SV40 large tumor antigen are physically associated with the same 54 kd cellular protein in transformed cells. Cell 1982;28:387–394.
Querido E, Marcellus RC, Lai A, et al. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J Virol 1997;71:3788–3798.
Querido E, Morrison MR, Chu-Pham-Dang H, et al. Identification of three functions of the adenovirus e4orf6 protein that mediate p53 degradation by the E4orf6-E1B55K complex. J Virol 2001;75:699–709.
Roth J, Konig C, Wienzek S, et al. Inactivation of p53 but not p73 by adenovirus type 5 E1B 55-kilodalton and E4 34-kilodalton oncoproteins. J Virol 1998;72:8510–8516.
Steegenga WT, Riteco N, Jochemsen AG, et al. The large E1B protein together with the E4orf6 protein target p53 for active degradation in adenovirus infected cells. Oncogene 1998;16:349–357.
Somasundaram K, El-Deiry WS . Inhibition of p53-mediated transactivation and cell cycle arrest by E1A through its p300/CBP-interacting region. Oncogene 1997;14:1047–1057.
Fuchs M, Gerber J, Drapkin R, et al. The p400 complex is an essential E1A transformation target. Cell 2001;106:297–307.
Lang SEHP . The adenovirus E1A oncoprotein recruits the cellular TRRAP/GCN5 histone acetyltransferase complex. Oncogene 2003;22:2836–2841.
Debbas M, White E . Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev 1993;7:546–554.
White E, Sabbatini P, Debbas M, et al. The 19-kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor alpha. Mol Cell Biol 1992;12:2570–2580.
Tollefson AE, Scaria A, Hermiston TW, et al. The adenovirus death protein (E3-11.6 K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J Virol 1996;70:2296–2306.
Sauthoff H, Pipiya T, Heitner S, et al. Late expression of p53 from a replicating adenovirus improves tumor cell killing and is more tumor cell specific than expression of the adenoviral death protein. Hum Gene Ther 2002;13:1859–1871.
Berk AJ . Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 2005;24:7673–7685.
Martin ME, Berk AJ . Adenovirus E1B 55 K represses p53 activation in vitro. J Virol 1998;72:3146–3154.
Levine AJ . The p53 protein and its interactions with the oncogene products of the small DNA tumor viruses. Virology 1990;177:419–426.
Koch P, Gatfield J, Lober C, et al. Efficient replication of adenovirus despite the overexpression of active and nondegradable p53. Cancer Res 2001;61:5941–5947.
Hobom U, Dobbelstein M . E1B-55-kilodalton protein is not required to block p53-induced transcription during adenovirus infection. J Virol 2004;78:7685–7697.
van Beusechem VW, van den Doel PB, Grill J, et al. Conditionally replicative adenovirus expressing p53 exhibits enhanced oncolytic potency. Cancer Res 2002;62:6165–6171.
Bergelson JM, Cunningham JA, Droguett G, et al. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997;275:1320–1323.
Wickham TJ, Mathias P, Cheresh DA, et al. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 1993;73:309–319.
Freimuth P . A human cell line selected for resistance to adenovirus infection has reduced levels of the virus receptor. J Virol 1996;70:4081–4085.
Pearson AS, Koch PE, Atkinson N, et al. Factors limiting adenovirus-mediated gene transfer into human lung and pancreatic cancer cell lines. Clin Cancer Res 1999;5:4208–4213.
Dmitriev I, Krasnykh V, Miller CR, et al. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J Virol 1998;72:9706–9713.
Krasnykh V, Dmitriev I, Navarro JG, et al. Advanced generation adenoviral vectors possess augmented gene transfer efficiency based upon coxsackie adenovirus receptor-independent cellular entry capacity. Cancer Res 2000;60:6784–6787.
Belousova N, Krendelchtchikova V, Curiel DT, et al. Modulation of adenovirus vector tropism via incorporation of polypeptide ligands into the fiber protein. J Virol 2002;76:8621–8631.
Belousova N, Korokhov N, Krendelshchikova V, et al. Genetically targeted adenovirus vector directed to CD40-expressing cells. J Virol 2003;77:11367–11377.
Everts M, Curiel DT . Transductional targeting of adenoviral cancer gene therapy. Curr Gene Ther 2004;4:337–346.
Wakimoto H, Fulci G, Tyminski E, et al. Altered expression of antiviral cytokine mRNAs associated with cyclophosphamide's enhancement of viral oncolysis. Gene Therapy 2004;11:214–223.
Graat HC, Wuisman PI, van Beusechem VW, et al. Coxsackievirus and adenovirus receptor expression on primary osteosarcoma specimens and implications for gene therapy with recombinant adenoviruses. Clin Cancer Res 2005;11:2445–2447; author reply 2447–2448.
Gu W, Ogose A, Kawashima H, et al. High-level expression of the coxsackievirus and adenovirus receptor messenger RNA in osteosarcoma, Ewing's sarcoma, and benign neurogenic tumors among musculoskeletal tumors. Clin Cancer Res 2004;10:3831–3838.
Witlox AM, Van Beusechem VW, Molenaar B, et al. Conditionally replicative adenovirus with tropism expanded towards integrins inhibits osteosarcoma tumor growth in vitro and in vivo. Clin Cancer Res 2004;10:61–67.
Glasgow JN, Kremer EJ, Hemminki A, et al. An adenovirus vector with a chimeric fiber derived from canine adenovirus type 2 displays novel tropism. Virology 2004;324:103–116.
Stoff-Khalili MA, Rivera AA, Glasgow JN, et al. A human adenoviral vector with a chimeric fiber from canine adenovirus type 1 results in novel expanded tropism for cancer gene therapy. Gene Therapy 2005;12:1696–1706.
Tsuruta Y, Pereboeva L, Glasgow JN, et al. Reovirus sigma1 fiber incorporated into adenovirus serotype 5 enhances infectivity via a CAR-independent pathway. Biochem Biophys Res Commun 2005;335:205–214.
Hedley SJ, Auf der Maur A, Hohn S, et al. An adenovirus vector with a chimeric fiber incorporating stabilized single chain antibody achieves targeted gene delivery. Gene Therapy 2006;13:88–94.
Modak S, Kramer K, Gultekin SH, et al. Monoclonal antibody 8H9 targets a novel cell surface antigen expressed by a wide spectrum of human solid tumors. Cancer Res 2001;61:4048–4054.
Onda M, Wang QC, Guo HF, et al. In vitro and in vivo cytotoxic activities of recombinant immunotoxin 8H9(Fv)-PE38 against breast cancer, osteosarcoma, and neuroblastoma. Cancer Res 2004;64:1419–1424.
Murphy A, Westwood JA, Teng MW, et al. Gene modification strategies to induce tumor immunity. Immunity 2005;22:403–414.
Bhowmick NA, Moses HL . Tumor–stroma interactions. Curr Opin Genet Dev 2005;15:97–101.
Chung LW, Baseman A, Assikis V, et al. Molecular insights into prostate cancer progression: the missing link of tumor microenvironment. J Urol 2005;173:10–20.
Condeelis J, Pollard JW . Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006;124:263–266.
Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005;121:335–348.
Pardoll D . Does the immune system see tumors as foreign or self? Annu Rev Immunol 2003;21:807–839.
Siegel PM, Massague J . Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer 2003;3:807–821.
Tlsty TD, Hein PW . Know thy neighbor: stromal cells can contribute oncogenic signals. Curr Opin Genet Dev 2001;11:54–59.
Dunn GP, Old LJ, Schreiber RD . The three Es of cancer immunoediting. Annu Rev Immunol 2004;22:329–360.
Khong HT, Restifo NP . Natural selection of tumor variants in the generation of ‘tumor escape’ phenotypes. Nat Immunol 2002;3:999–1005.
Zou W . Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer 2005;5:263–274.
Jooss K, Chirmule N . Immunity to adenovirus and adeno-associated viral vectors: implications for gene therapy. Gene Therapy 2003;10:955–963.
Schagen FH, Ossevoort M, Toes RE, et al. Immune responses against adenoviral vectors and their transgene products: a review of strategies for evasion. Crit Rev Oncol Hematol 2004;50:51–70.
Ikeda K, Ichikawa T, Wakimoto H, et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med 1999;5:881–887.
Wakimoto H, Johnson PR, Knipe DM, et al. Effects of innate immunity on herpes simplex virus and its ability to kill tumor cells. Gene Therapy 2003;10:983–990.
MacEwen EG . Spontaneous tumors in dogs and cats: models for the study of cancer biology and treatment. Cancer Metastasis Rev 1990;9:125–136.
Withrow SJ, Powers BE, Straw RC, et al. Comparative aspects of osteosarcoma. Dog versus man. Clin Orthop Relat Res 1991;270:159–168.
Vail DM, MacEwen EG . Spontaneously occurring tumors of companion animals as models for human cancer. Cancer Invest 2000;18:781–792.
Hemminki A, Kanerva A, Kremer EJ, et al. A canine conditionally replicating adenovirus for evaluating oncolytic virotherapy in a syngeneic animal model. Mol Ther 2003;7:163–173.
Ternovoi VV, Le LP, Belousova N, et al. Productive replication of human adenovirus type 5 in canine cells. J Virol 2005;79:1308–1311.
Acknowledgements
This work was funded in part by Grants CA93796, CA98543 and AR46031 from the National Institutes of Health, Bethesda, MD and the Haley's Hope Memorial Support Fund for Osteosarcoma Research, University of Alabama at Birmingham. We thank Drs Joel Glasgow and Justin Roth for their thorough review and careful critique of the manuscript. We also thank Dr Natalya Belousova for her help in the preparation of this submission.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ternovoi, V., Curiel, D., Smith, B. et al. Adenovirus-mediated p53 tumor suppressor gene therapy of osteosarcoma. Lab Invest 86, 748–766 (2006). https://doi.org/10.1038/labinvest.3700444
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700444
Keywords
This article is cited by
-
The expression and significance of IDH1 and p53 in osteosarcoma
Journal of Experimental & Clinical Cancer Research (2010)
-
Establishment of ponasterone A-inducible the wild-type p53 protein-expressing clones from HSC-1 cells, cell growth suppression by p53 expression and the suppression mechanism
Archives of Dermatological Research (2009)
