
Abstract
A comprehensive genomic analysis of single cells is needed for numerous scenarios in tumor genetics, clinical diagnostics and forensic application. PCR protocols were developed which allow an unbiased amplification of the whole genome of a single cell for subsequent analyses by comparative genomic hybridization (CGH). However, verification of single-cell CGH results has been impossible as the procedure naturally involves the destruction of the respective cell. Here we show that the genome of individual cells can be analyzed by two different single cell techniques applied sequentially to the same cell. In a first step, interphase fluorescence in situ hybridization (FISH) is applied. After evaluation of the interphase-FISH signals, cells of interest can be selected for a further analysis. Single cells are collected by laser microdissection, the DNA is amplified by linker-adaptor PCR and subjected to CGH-analysis. This strategy offers new opportunities for a sophisticated selection of cells based on interphase-FISH signals. Furthermore, the sequential application of two different single-cell approaches to the same single-cell represents the only option to control and verify the single-cell CGH results. We demonstrate the feasibility of this approach with a series of experiments including cells from pre- and postnatal diagnostics, for example, cells with trisomies 13, 18, or 21, respectively, leukemia and tumor cells and tissue sections.
Similar content being viewed by others

Main
There is an increasing demand for comprehensive genome analyses of single cells. In cancer genetics, single-cell approaches allow monitoring minimal residual disease or the assessment of heterogeneity within a primary tumor. In clinical diagnostics single-cell analyses are important in cases of mosaicism. In addition, advanced single-cell analyses may pave the way for noninvasive prenatal diagnostic strategies using fetal cells derived from maternal blood. Furthermore, whole genome single-cell analyses are instrumental in forensic application.
For a screening of copy number changes within the entire genome of single cells we and others developed protocols which allow the unbiased amplification of the DNA of single cells for subsequent comparative genomic hybridization (CGH) experiments.1, 2, 3 However, it is difficult to verify the results of single-cell CGH as the respective cell is destroyed during the procedure and therefore unavailable for further experiments.
Here we show that our CGH protocol1 can be adapted for the use of single cells collected by microdissection and laser catapulting. Furthermore, we reasoned that the only possible strategy to verify and control single-cell CGH results should consist in a sequential double-analysis using two different single-cell approaches to the same cell. In a first step, multicolor interphase fluorescence in situ hybridization (FISH) is employed and the interphase signals are evaluated. In a second step, cells are microdissected and reanalyzed after PCR-amplification of the genome by single-cell CGH. This sequence interphase FISH first followed by single-cell CGH should in addition greatly facilitate the selection of cells for subsequent detailed single-cell CGH.
To test our approach we conducted a series of experiments with normal cells from peripheral blood or from umbilical cord blood, clinical cases with trisomies of chromosomes 13, 18, or 21, respectively, renal cancer and colorectal tumor cell lines and leukemia cases. Furthermore, we tested whether our approach is also applicable to tissue sections after interphase-FISH.
Materials and methods
Cells and Cell Culture
We used a variety of normal cells, cells from clinical diagnostics, leukemia cases and tumor cell lines and tissue sections. Normal cells were derived from peripheral blood or from umbilical cord blood. In addition, we used cells from patients with acute myeloid leukemia (AML) and acute lymphocytic leukemia (ALL), in which standard cytogenetic procedures had shown normal chromosomal compositions. From our own diagnostic cytogenetic laboratory we received cells with trisomies 13, 18, or 21, respectively. Blood was diluted 1:1 with Hanks solution (Sigma). The nucleated cells were isolated by single-density centrifugation using Ficoll-Paque (1.077 g/ml; Sigma) according to manufacturer's instructions.
The cell lines HCT116 and LOVO were generously provided by Dr Christoph Lengauer (Johns Hopkins Oncology Center, Baltimore, MD, USA). Both HCT116 and LOVO are known to have microsatellite instability and are chromosomally stable.4 The karyotype of HCT116 has been described by us and others as: 45,X,-Y,der(10)dup(10)(q24q26)t(10;16)(q26;q24),der(16)t(8;16)(q13;p13),der(18)t(17;18)(q21;p11.3).5, 6 We verified the karyotype prior to our experiments.
The karyotype of LOVO has previously been described as 49,XY,+5,+7,+der(12)t(2;12),i(15)(q10).5 When we karyotyped our cell line with the designation ‘LOVO’ we found the karyotype 47,XY,t(2;12),+7 resulting in an over-representation only of chromosome 7. In addition, we found in all cells one or two double minute chromosomes. Culturing of cells was as described.6
The renal cell cancer cell line 769P was kindly provided by Dr NP Carter (Wellcome Trust Sanger Institute, Cambridge, UK). The karyotype was analyzed by multiplex-FISH (M-FISH). Most cells have a hypotetraploid chromosome number (n=81–83) and there was cell-to-cell variability. However, karyotyping suggested that some numerical aberrations should be present in each cell. In all cells we found unbalanced translocations between chromosome 1 and 9 resulting in over-representations of 1q. Furthermore, each cell had one or two copies of a der(11)t(8;11), which causes an over-representation of 8q and loss of distal 11q material. In addition, we observed in all cells an under-representation of chromosome 14. These four regions were used to determine the accuracy of cell-pool or single-cell CGH. Other gains or losses, for example, loss of chromosome 6, were observed in only a subset of cells.
Preparation of Cells on Membrane-Coated Slides
Cell suspensions were centrifugated at 700 g for 10 min and the cell pellet was washed twice with PBS. In all, 200 μl of cell solution containing approximately 500 000 cells was transferred onto a membrane covered microscope slide by cytocentrifugation at 1000 rpm for 3 min. According to the manufacture's instructions (PALM Microlaser Technologies, Bernried, Germany), cells can be transferred to two different membranes. One is a polyester membrane (POL), the second membrane is made of polyethylene-naphtalate (PEN). We never obtained a PCR-product using the POL-membrane. In contrast, cells on the PEN-membrane yielded usable PCR amplification products. Hence, all experiments were carried out with the PEN-membrane.
Treatment of Membrane-Coated Slides Prior to Hybridization
Cytospins were incubated in a 0.5% formaldehyde/PBS solution at room temperature for 10 min and washed 3 × 3 min with 1 × PBS followed by a fixation step in a freshly prepared cold mixture of 3:1 methanol:acetic acid at 4°C for 30 min. Subsequently, cells were treated with 0.1% Triton X at ambient temperature for 15 min and digested for 10–15 min at 37°C using 60 μl pepsin [10 mg/ml] in 50 ml 0.1 M hydrochloric acid. Slides were washed 2 × 5 min in 1 × PBS, dehydrated with ethanol, air dried and denatured in a solution containing 2 × SSC and 70% formamide, pH 7.0, at 72°C for 2 min. The hybridization probe was dropped onto the slide, covered, sealed with rubber cement and incubated in a moist chamber at 37°C for 2 nights.
Hybridization and Evaluation of Painting and Chromosome Centromere-Specific Probe, M-FISH
Painting and chromosome centromere-specific probes were hybridized according to standard protocols as published previously.7, 8 In brief, we used custom made probe sets consisting of various combinations of centromere probes for the following chromosomes: seven (PZ7.6B), eight (PZ8.4), 11 (PRB11), 17 (PZ17-14), and 18 (2Xba). These centromere probes were generously provided by Dr M Rocchi (for detailed information see http://www.biologia.uniba.it/rmc/index.html). We applied painting probes for chromosomes 3 and 6, which were made by flow cytometry and kindly provided by Dr NP Carter (Wellcome Trust Sanger Institute, Cambridge, UK). In some instances we used commercially available probe sets for centromere probes for chromosomes 18, X, Y or region-specific probes for chromosomes 13 and 21 (Abbott-Vysis, Aneu Vysion Kit). Table 1 provides an overview, which probe sets were hybridized to the respective cell systems.
M-FISH was performed according to our previously published protocols9, 10 using seven different fluorochromes to improve resolution.11
Isolation of Cells by Laser Microdissection and Pressure Catapulting
Cells were isolated by the PALM MicroBeam System (PALM Microlaser Technologies, Bernried, Germany;12, 13, 14). Isolated cells were lifted into the cap of a 200 μl Eppendorf tube containing 4.5 μl digestion-mix. Collected material was centrifuged into the tube at 13 000 rpm for 15 min (Mikro 22R, Hettich, Germany).
Single-Cell Whole Genome Amplification by PCR
Single-cell whole genome amplification was performed based on a previously published protocol1 with modifications as described in Thalhammer et al.14 In brief, isolated material was digested in 4.5 μl of proteinase K digestion buffer (0.5 μl of 10 × One-Phor-All-Buffer-Plus (Amersham Pharmacia Biotech), 0.13 μl 10% Tween 20 (Sigma, Germany), 0.13 μl 10% Igepal (Sigma), 0.13 μl Proteinase K (10 mg/ml, Sigma)) for 15 h at 42°C in an MJ-Research PTC-200 thermocycler (Waltham, USA). Proteinase K was inactivated at 80°C for 10 min. MseI restriction endonuclease digest was performed in 5 μl by adding 0.25 μl of MseI (50.000 U/μl, New England Biolabs, Germany) and 0.25 μl H20 for 3 h at 37°C with subsequent inactivation at 65°C for 5 min. Preannealing of adaptors was achieved by adding Lib1 primer (5′-AGT GGG ATT CCT GCT GTC AGT-3′) and ddMse 11 (5′-TAA CTG ACA GCdd-3′), 0.5 μl each of 100 μM stock solution, 0.5 μl One- Phor-All-Buffer and 1.5 μl of H2O. Annealing was started at 65°C and was shift down to 15°C with a ramp of 1°C/min. At 15°C, 1 μl of ATP (10 mM) and 1 μl T4-DNA-Ligase (5 U/μl, Roche, Germany) were added and incubated overnight at 15°C.
For primary amplification 40 μl consisting of 3 μl of BM Puffer 1 (Expand long template, Roche), 2 μl of dNTPs (10 mM), 1 μl Pol- Mix (3.5 U/μl; Roche) and 35 μl H2O were added to the ligation product.
The PCR program started with 68°C for 3 min and was subsequently programmed to 94°C (40 s), 57°C (30 s) and 68°C (1 min 30 s) (ramp+1 s/cycle) for 14 cycles, 94°C (40 s), 57°C (30 s) (ramp+1°C/cycle) and 68°C (1 min 45 s) (ramp+1 s/cycle) for eight cycles; 94°C (40 s), 65°C (30 s) and 68°C (1 min 53 s) (ramp+1 s/cycle) for 22 cycles followed by a final elongation step at 68°C for 3 min 40 s.
In all, 2 μl of primary DNA were reamplified in a final volume of 50 μl using 5 μl BM Buffer 2 (Roche, Germany), 5.5 mM MgCl2 (Gibco, Germany), 0.2 mM dNTP (Roche, Germany), 4 μM Lib1 primer, 2.5 U/μl Taq-polymerase (Roche, Germany). After a denaturation step of 95°C (10 min), 45 cycles were programmed to 95°C (30 s), 50°C (30 s), 72°C (2 min) and a final elongation step with 72°C (7 min).15 The PCR-reamplification products were labeled by standard nicktranslation.
Chromosome-CGH
Metaphase preparation and CGH-hybridization were performed as published.16
Labeled amplification product of one or several cells was mixed with biotin-labeled reference DNA (derived from placenta), Cot-1 DNA and salmon sperm DNA. The hybridization mix was denatured at 78°C for 7 min and hybridized to metaphase spreads.
For detection of hybridization signals of test DNA/control DNA sheep anti-dig FITC (200 μg; Roche)/Avidin Cy3.5 (1 mg/ml; Rockland) were used.
Image Acquisition and Evaluation
Images were captured with a Leica DMRXA-RF8 microscope equipped with a Sensys charge-coupled device (CCD) camera (Photometrics; Kodak KAF 1400 chip). The Leica microscope and corresponding camera were controlled by the Leica QFISH software (Leica Microsystems Imaging Solutions, Cambridge, UK). Preceding laser microdissection signals were visualized and documented with a Sony DXC-390P 3CCD camera, which was connected to the PALM MicroBeam System.
Quantitative evaluation of the ratio of test DNA and control DNA was performed using the Leica Q-CGH-program.
Results
An overview of our experiments is shown in Table 1. We used cell systems with a known normal chromosome count (normal female blood, male umbilical cord blood), cells from clinical diagnostics with known trisomies for chromosomes 21, 13, and 18, cells from patients with AML and ALL, colorectal cancer cell lines (HCT116 and LOVO), a renal cell cancer cell line (796P) and a kryosection from a tumor of a colon cancer patient. With the exception of the tissue section we performed for each cell system the following set of experiments: CGH was carried out in the conventional way, that is, with DNA extracted from a large number of cells. In addition, we performed CGH with a pool of cells. A ‘pool’ refers to a cell number of 5–10 microdissected cells, which were collectively transferred to the same tube for further processing. These pool-experiments were performed with and without previous FISH to estimate the impact of the FISH on the outcome of the CGH-profile. The same set of experiments was also performed with single cells (Table 1).
The experimental schematic outline is illustrated in Figure 1. Exemplary interphase-FISH signals and microdissection of the respective single cells are depicted in Figure 2. To test the feasibility and the robustness of this approach we addressed two questions. First, is the accuracy of the CGH result critically dependent on the number of cells used to extract DNA? Second, does a previously performed interphase-FISH experiment to the same cell have an effect on the outcome? To evaluate the CGH-profiles we used an accuracy rate. The accuracy rate provides the percentage of experiments, which identified all numerical aberrations correctly and in which no additional, unexpected numerical aberrations were observed. This accuracy rate could be estimated because we used very stable cell systems with no or a very low heterogeneity and with known numerical aberrations. Furthermore, the accuracy rate was derived by comparisons with the CGH profiles obtained after conventional CGH, with DNA from cell pools and with DNA from a single cell.

Experimental schematic outline of our sequential application of two different single-cell approaches to the same cell. In a first step, multicolor interphase-FISH is applied and the interphase signals are evaluated. In this example, the cell in the center has three green signals while all other cells have two signals. The presence of an over-representation can be verified in a second step. To this end, the cell is laser microdissected and catapulted into a tube. The DNA of the cell is amplified using a linker-adaptor approach employing restriction enzyme digestion with MseI, an adaptor preannealing, ligation, primary PCR, reamplification (secondary PCR) and labeling. Finally, the amplification product can be analyzed for gains and losses, either by chromosome CGH or by array-/matrix-CGH.

Visualization of interphase-FISH signals prior to the subsequent processing with single-cell CGH shown both for a normal male cell (a–d) and a cell carrying chromosomal imbalance (e–h). (a and e) The FISH images are shown slightly enlarged for better visibility of the interphase signals. In (a) one signal each for the X- (green) and the Y-chromosome (red) channel are visible. This cell corresponds to the CGH-profile shown in Figure 3c. Three signals for the chromosome 18 centromere probe are detectable in the cell shown in (e). The subsequent CGH-profile is depicted in Figure 4c. (b and f) The same cells are visualized in phase contrast. The shape of each cell is clearly discernible (arrows), no other cells are in the vicinity. The cutting edge for the subsequent microdissection can be determined on the computer screen (blue circle). A scale bar is shown in each image. (c and g) The membrane is almost completely cut. (d and h) The respective areas after microdissection showing the hole in the membrane after microdissection.
CGH with DNA Derived from Cell Pools or Single Cells without FISH
The purpose of these experiments was to answer the first question mentioned above, that is, to explore whether the number of cells selected for DNA-amplification by linker-adaptor PCR and subsequent CGH experiments has a significant impact on the accuracy of the results.
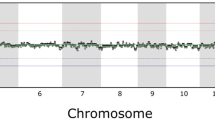
Initial experiments were performed with normal male and female cells or cells with one additional chromosome (trisomy 13, 18, 21). We correctly identified the sex of the donor of the respective cell and the over-represented chromosomes in all conducted experiments (Table 1). Examples for ratio profiles of male umbilical cord blood and a case with trisomy 18 are shown in Figures 3a, b and 4a, b.

CGH profiles obtained with DNA from male umbilical cord blood cells. (a) CGH-profile obtained with DNA from a cell pool consisting of 10 cells. (b) Single-cell CGH-profile without FISH. (c) CGH-profile from a single cell on which previously centromere probes for chromosomes 18, X, and Y had been hybridized (the signals for the X- and the Y-chromosomes are shown in Figure 1a).

CGH results derived from cells of a female with a known trisomy 18. (a) CGH-profile derived from a cell pool of five cells. (b) Single-cell CGH-profile of a trisomy 18 cell without a previous FISH experiment. (c) Single-cell CGH-profile of a trisomy 18 cell after a previous FISH experiment with centromere probes for chromosomes 18, X and Y (the signals for the chromosome 18 probe are depicted in Figure 2e).
In a next step we analyzed one case each of AML and ALL. In each case standard cytogenetic banding analysis and M-FISH indicated a normal karyotype and neither conventional nor single-cell CGH were suggestive for the presence of any clones with some imbalances (Figure 5).

CGH-profile derived from a single AML cell on which previously a painting probe for chromosome 6 had been hybridized. Interphase-FISH had identified two copies of chromosome 6 and the CGH-profile confirms the balanced copy number.
Additional tests were performed with the colorectal cancer cell lines HCT116 and LOVO and with the renal cancer cell line 769P. In each case the obtained conventional CGH-profiles were similar to the results with cell-pool DNA or DNA from single cells (data not shown).
The outcome of all experiments is summarized in Table 1. In the vast majority of cases we obtained an accuracy rate of 100%. Thus, we conclude that using our protocols DNA from single cells can be analyzed by CGH with the same accuracy as DNA derived from cell pools or large cell numbers.
CGH with DNA Derived from Cell Pools or Single Cells after FISH
The purpose of these experiments was to answer the above-mentioned second question, that is, whether a previously performed interphase-FISH experiment to the same cell has an effect on the accuracy of the CGH ratio profile.
Therefore, the same set of experiments as outlined above was repeated after various DNA probes as listed in Table 1 were hybridized to the respective cells and evaluated. In the interphase-FISH experiments we did not use the DAPI counterstain and minimized exposure of cells to UV-light as this may result in a fragmentation of DNA.17
The selection and microdissection of a single cell after a FISH experiment is illustrated in Figure 1. The corresponding single-cell CGH-profile to Figure 2a–d, which was obtained after this FISH experiment is shown in Figure 3c. Similarly, Figure 2e–h shows the interphase-FISH image related to the CGH-profile shown in Figure 4c. From these profiles and from other experiments (own unpublished data) it is apparent that the previous FISH experiment does not alter the expected CGH profile.
Finally, we asked whether our approach would be applicable to tissue sections. We used a tissue section (5 μm) from a colorectal cancer to explore this question. Small areas of the tissue, consisting of about 50–100 cells, were microdissected without FISH and after FISH with centromere probes for chromosomes 7, 11, and 17. The two resulting CGH-profiles identified in each case regions, which were commonly gained or lost (eg loss of 1p, 4q, 5q, 18q and gain of 7p, 8q, 20) suggesting that our approach can be extended to tissue sections.
Table 1 lists the accuracy rate for cell-pool and single-cell CGH experiments performed after interphase-FISH. An accuracy rate could not be given for the tissue section experiments, as no base-line values for comparison were available.
The Interphase-FISH DNA Probes do not Affect the CGH-Profile
An important question was, whether a hybridized probe may affect the single cell CGH-profile. A successful hybridization should result in an over-representation of the target sequence, as this region is present in the respective cell as both the cellular DNA and the probe DNA. Hence, a hybridized probe may increase the amount of the respective chromosomal region and may therefore be visible as a gain in the CGH-profile.
This concern should not apply if centromere probes were hybridized as in CGH experiments hybridization to centromeres is suppressed by the addition of an excess of unlabeled Cot1-DNA. However, as illustrated in our figures, even the hybridization of chromosome-specific probes or of other chromosome region-specific probes did not affect the CGH-profile. For example, Figure 5 illustrates the CGH-profile of a single AML cell on which previously a chromosome 6 painting probe had been hybridized. The CGH-profile shows a balanced ratio value for the entire chromosome 6, which corresponds to the results obtained by banding analysis and M-FISH.
Therefore, we tested whether hybridized probes are not co-amplified by the linker-adaptor PCR. To this end we tried to amplify labeled DNA-probes with our linker-adaptor PCR-approach, however, we never obtained an amplification product. Thus, the incorporated hapten or fluorochrome may inhibit the amplification by linker–adaptor PCR. Alternatively, the incorporated hapten or fluorochrome may interfere with the MseI restriction enzyme. In any case, after FISH the linker-adaptor PCR amplifies only efficiently the cellular DNA but not the previously hybridized probe. Therefore, a previous FISH-experiment does not affect the CGH-profile.
The Sequential Single-Cell Analyses are not Especially Susceptible to DNA Contamination
The risk of DNA contamination is an especial problem for single-cell PCR procedures. In our case the cells examined are exposed to potential contaminants in the laboratory for extended periods of time. However, under the conditions used in our laboratory none of the CGH-profiles, which we obtained from cells with known aneuploidies, suggested the presence of a possible contamination, which should have been visible as an unexpected deviation from the CGH-profile. To further verify that our procedure is not especially susceptible to a significant contamination, we microdissected several regions (n=10), which consisted of membrane only without any cells. These microdissected membranes were subjected to the same PCR amplification procedure as our single cells. We did not get an amplification product in any of these experiments, suggesting that our precautions are sufficient to reduce the risk of contamination to a minimum.
Discussion
We used clinical cases and tumor cell lines with known copy number changes, which should ideally be suited to test the accuracy rate of single-cell CGH. Our procedure is to our knowledge the first implementation of successful single-cell CGH after interphase-FISH.
In our approach cells were isolated by laser microdissection and pressure catapulting. The advantages of microdissection as compared to micromanipulation include that the cell selection procedure requires less manual skills of the operator and the contact-free transfer of cells into a tube. Furthermore, as shown here, it allows the application of additional single-cell approaches, such as interphase-FISH.
Even if the cells were exposed to a previous interphase-FISH experiment, we generally obtained relatively straight ratio profiles, as shown in our figures, which are easy to interpret and which reduce ambiguous ratio values. This represents a significant difference to other published single-cell CGH procedures, which are modifications of the DOP-PCR approach and usually result in a zigzag shape of the ratio curve as visible in the respective publications.2, 3, 18, 19, 20 The high quality of our ratio profiles is probably a reason why the accuracy rate was generally in the range of close to 100%. Therefore, we could use our single-cell CGH-approach already for monitoring minimal residual disease albeit without FISH,13 and we can now apply our strategy for diagnostic applications (own unpublished data).
The sequential application of two different single-cell techniques increases the accuracy of single-cell analysis. Neither interphase-FISH alone nor single-cell CGH alone is 100% accurate. However, the copy number of a chromosomal region can be considered as accurately established, if both tests yield concordant results. In fact, the comparison of the results from a multicolor-interphase FISH assay with the subsequent CGH-profile offers the only opportunity to control results obtained with either method as both results are derived from the same cell. In the case of discrepant results, for example, that FISH shows one signal and subsequent CGH two copies, it remains uncertain which result is correct. Therefore, these cells should be excluded from further analysis, which should improve the overall result.
Another advantage of the combined technique is the option to select cells for single-cell CGH based on FISH signals, which will increase the efficiency of single-cell analyses. For example, an important application represents the analysis of disseminated epithelial cells in the bone marrow of patients with cancer. These cells may have no or a various number of copy number changes.13, 21, 22 A preceding interphase-FISH analysis offers the opportunity for the sophisticated selection of cells for further analysis based on certain interphase-FISH patterns.
The major limitation of chromosome-CGH is the poor resolution, which has been estimated to be in the range of 10 Mb.23 Resolution could be improved with matrix-/array-CGH.24, 25 However, up to date, single-cell DNA amplification products could not successfully be applied to arrays. First attempts to hybridize our single-cell PCR-amplification products to a 1 Mb array26 resulted in a heterogeneous hybridization pattern (H Fiegler, N Carter, The Wellcome Trust Sanger Institute, Cambridge, UK, personal communication). Similar results were also reported from others (P Lichter, German Cancer Research Center, Heidelberg, Germany, personal communication). Efforts are underway to refine the protocol to apply the amplification product to arrays.
The protocols described here, will improve the genomic analysis of single cells. This will contribute to a better understanding of many biologic processes and causes of disease at a cellular level.
References
Klein CA, Schmidt-Kittler O, Schardt JA, et al. Comparative genomic hybridization, loss of heterozygosity, and DNA sequence analysis of single cells. Proc Natl Acad Sci USA 1999;96:4494–4499.
Wells D, Sherlock JK, Handyside AH, et al. Detailed chromosomal and molecular genetic analysis of single cells by whole genome amplification and comparative genomic hybridisation. Nucleic Acids Res 1999;27:1214–1218.
Voullaire L, Wilton L, Slater H, et al. Detection of aneuploidy in single cells using comparative genomic hybridization. Prenat Diagnosis 1999;19:846–851.
Lengauer C, Kinzler KW, Vogelstein B . Genetic instability in colorectal cancers. Nature 1997;386:623–627.
Abdel-Rahman WM, Katsura K, Rens W, et al. Spectral karyotyping suggests additional subsets of colorectal cancers characterized by pattern of chromosome rearrangement. Proc Natl Acad Sci USA 2001;98:2538–2543.
Bunz F, Fauth C, Speicher MR, et al. Targeted inactivation of p53 in human cells does not result in aneuploidy. Cancer Res 2002;62:1129–1133.
Uhrig S, Schuffenhauer S, Fauth C, et al. Multiplex-FISH (M-FISH) for pre- and postnatal diagnostic applications. Am J Hum Genet 1999;65:448–462.
Maierhofer C, Gangnus R, Diebold J, et al. Multicolor deconvolution microscopy of thick biological specimens. Am J Path 2003;162:373–379.
Speicher MR, Ballard SG, Ward DC . Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat Genet 1996;12:368–375.
Eils R, Uhrig S, Saracoglu K, et al. An optimized, fully automated system for fast and accurate identification of chromosomal rearrangements by multiplex-FISH (M-FISH). Cytogenet Cell Genet 1998;82:160–171.
Azofeifa J, Fauth C, Kraus J, et al. An optimized probe set for the detection of small interchromosomal aberrations by 24-color FISH. Am J Hum Genet 2000;66:1684–1688.
Schütze K, Lahr G . Identification of expressed genes by laser-mediated manipulation of single cells. Nat Biotechnol 1998;16:737–742.
Gangnus R, Langer S, Breit S, et al. Genomic profiling of viable and proliferative micrometastatic cells from early stage breast cancer patients. Clin Cancer Res 2004;10:3457–3464.
Thalhammer S, Langer S, Speicher MR, et al. Generation of chromosome painting probes from single chromosomes by laser microdissection and linker-adaptor PCR. Chrom Res 2004;12:337–343.
Snijders AM, Nowak N, Segraves R, et al. Assembly of microarrays for genome-wide measurement of DNA copy number. Nat Genet 2001;29:263–264.
du Manoir S, Speicher MR, Joos S, et al. Detection of complete and partial chromosome gains and losses by comparative genomic in situ hybridization. Hum Genet 1993;90:590–610.
Weimer J, Kiechle M, Arnold N . FISH-microdissection (FISH-MD) analysis of complex chromosome rearrangements. Cytogenet Cell Genet 2000;88:114–118.
Wilton L, Williamson R, McBain J, et al. Birth of a healthy infant after preimplantation confirmation of euploidy by comparative genomic hybridization. New Eng J Med 2001;345:1537–1541.
Wells D, Escudero T, Levy B, et al. First clinical application of comparative genomic hybridization and polar body testing for preimplantation genetic diagnosis of aneuploidy. Fertil Steril 2002;78:543–549.
Wilton L, Voullaire L, Sargeant P, et al. Preimplantation aneuploidy screening using comparative genomic hybridization of fluorescence in situ hybridization of embryos from patients with recurrent implantation failure. Fertil Steril 2003;80:860–870.
Klein CA, Blankenstein TJ, Schmidt-Kittler O, et al. Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet 2002;360:683–689.
Schmidt-Kittler O, Ragg T, Daskalakis A, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci USA 2003;100:7737–7742.
Bentz M, Plesch A, Stilgenbauer S, et al. Minimal sizes of deletions detected by comparative genomic hybridization. Genes Chrom Cancer 1998;21:172–175.
Solinas-Toldo S, Lampel S, Stilgenbauer S, et al. Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chrom Cancer 1997;20:399–407.
Pinkel D, Segraves R, Sudar D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 1998;20:207–211.
Fiegler H, Carr P, Douglas EJ, et al. DNA microarrays for comparative genomic hybridization based on DOP-PCR amplification of BAC and PAC clones. Genes Chromosome Cancer 2003;36:361–374.
Acknowledgements
We are grateful to Gaby Lederer, Corinna Keri, and Doris Sollacher for expert technical assistance. The leukemia samples were kindly provided by Dr Georg Ledderose (Klinikum Großhadern, Munich, Germany) and the colon tissue sections by Dr Stefan Seidl (Technical University Munich, Germany). We appreciate the helpful comments and suggestions from Julie Waldispühl regarding Figure 1. This work was supported by the Wilhelm Sander-Stiftung, the Deutsche Forschungsgemeinschaft (Sp 460/5-1) and the Bundesministerium für Bildung und Forschung (BMBF, NGFN KB PO6T5).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Langer, S., Geigl, J., Gangnus, R. et al. Sequential application of interphase-FISH and CGH to single cells. Lab Invest 85, 582–592 (2005). https://doi.org/10.1038/labinvest.3700249
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700249
Keywords
This article is cited by
-
Methodology for Y Chromosome Capture: A complete genome sequence of Y chromosome using flow cytometry, laser microdissection and magnetic streptavidin-beads
Scientific Reports (2018)
-
A novel single cell method to identify the genetic composition at a single nuclear body
Scientific Reports (2016)
-
Amplification of multiple genomic loci from single cells isolated by laser micro-dissection of tissues
BMC Biotechnology (2008)
-
Neue Verfahren für Einzelzellanalysen in Forschung und Diagnostik
Medizinische Genetik (2008)
-
Single-cell isolation from cell suspensions and whole genome amplification from single cells to provide templates for CGH analysis
Nature Protocols (2007)
