
Abstract
Various types of mutations exist that exert an effect on the normal function of a gene. Among these, exon/gene deletions often remain unnoticed in initial mutation screening. Until recently, no fast and efficient methods were available to detect this type of mutation. Molecular detection methods for gene copy number changes included Southern blot (SB) and fluorescence in situ hybridisation, both with their own intrinsic limitations. In this paper, we report the development and application of a fast, sensitive and high-resolution method for the detection of single exon or larger deletions in the VHL gene based on real-time quantitative PCR (Q-PCR). These deletions account for approximately one-fifth of all patients with the von Hippel–Lindau syndrome, a dominantly inherited highly penetrant familial cancer syndrome predisposing to specific malignancies including phaeochromocytomas and haemangioblastomas. Our VHL exon quantification strategy is based on SYBR Green I detection and normalisation using two reference genes with a normal copy number, that is, ZNF80 (3q13.31) and GPR15 (3q12.1). Choice of primer sequences and the use of two reference genes appears to be critical for accurate discrimination between 1 and 2 exon copies. In a blind Q-PCR study of 29 samples, all 14 deletions were detected, which is in perfect agreement with previously determined SB results. We propose Q-PCR as the method of choice for fast (within 3.5 h), accurate and sensitive (ng amount of input DNA) exon deletion screening in routine DNA diagnosis of VHL disease. Similar assays can be designed for deletion screening in other genetic disorders.
Similar content being viewed by others


Main
Deletions or duplications encompassing single or multiexons, entire genes or larger chromosomal regions are a frequent cause of human genetic disorders. Recurrent microdeletions and segmental duplications are typically detected by locus-specific probes hybridising to the affected chromosomal regions. For exon deletions, however, sensitivity of fluorescence in situ hybridisation (FISH) is too low and other methods are required. In this study, we propose a promising alternative for former gene copy number quantification methods, applied for the detection of germline small exon or larger VHL gene deletions predisposing to the von Hippel–Lindau (VHL) syndrome. Different mutational mechanisms inactivate the VHL gene, that is, small intragenic mutations, and deletions (including deletion of large parts or even of the entire VHL gene), and somatic inactivation by hypermethylation of the promoter region.1, 2 In 20–30% of the VHL families, large (exon or entire gene) deletions were observed;3, 4, 5 in only a minority of these patients, a total VHL gene deletion is found (15% in our cohort of patients fulfilling the clinical criteria for VHL). Updated summaries of VHL germline mutations can be found on the World Wide Web at http://www.umd.necker.fr/, at http://uwcmml1s.uwcm.ac.uk/uwcm/mg/search/120488.html6 and at http://web.ncifcrf.gov/research/kidney/vhlcor.html. VHL germline deletions have been previously demonstrated by Southern blot analysis (SB),4 occasionally in combination with pulse field gel electrophoresis,7, 8 and FISH.9, 10, 11 Each of the above-mentioned methods, however, has its own intrinsic disadvantages. While it has been shown that SB analysis using a control probe is a sensitive method for detecting deletions encompassing the VHL gene,4 this technique requires large amounts of high-quality DNA, is laborious, time-consuming and often employs the use of hazardous radioisotopes. FISH allows the assessment of VHL loss at single-cell resolution, but has a limited sensitivity for deletions smaller than approximately 10 kb. More recently, PCR-based alternative strategies have been proposed for detection of exon or entire gene deletions, that is, long-range PCR,12 multiplex-amplifiable probe hybridisation,13 multiplex ligation probe-dependent Amplification (MLPA)14 and real-time quantitative PCR (Q-PCR).15 Here, we describe the development and extensive evaluation of a deletion screening method based on real-time Q-PCR to detect VHL exon deletions. Our approach is both fast, accurate, sensitive and specific, making it the method of choice for VHL and other gene deletion screening diagnostic tests.
Materials and methods
Patient Samples
Blood samples were obtained from 17 individuals from 15 unrelated families, representing patients with clinical features suggesting VHL disease (ie in the presence of a positive familial history, a diagnosis of VHL disease can be made by the identification of a single retinal or cerebellar haemangioblastoma, renal cell carcinoma or phaeochromocytoma, in an at-risk individual;16 in the absence of a VHL family history, two or more haemangioblastoma, or haemangioblastoma combined with a further typical VHL tumour are required17). Patient 17 (Table 2) does not fulfil the above requirements. This patient was referred for VHL mutation screening by an internal medicine department (Utrecht, The Netherlands), in the context of a diagnosis of phaeochromocytoma at a young age. Other VHL-related clinical symptoms were not reported, neither was there a positive family history for the disease. No germline mutation could be identified in the VHL gene or in other phaeochromocytoma-associated disease genes (RET, SDHB, SDHD) in this patient. The VHL patients were mainly selected for having a (partial or entire) VHL gene deletion (14/17), as determined previously by SB (see Table 2). High molecular weight DNA was isolated from peripheral blood samples using the QIAamp DNA Blood mini kit following the instructions of the manufacturer (Qiagen, Hilden, Germany) or by an established desalting method. Constitutional DNA from a VHL patient with an entire VHL gene deletion and neuroblastoma cell line SJNB-6 demonstrating distal 3p loss (including the VHL locus) was used as a positive control for deletion in SB and real-time Q-PCR, respectively. In addition, 24 normal diploid control samples from healthy individuals were included in the analysis (12 samples for VHL exon deletion screening, and 24 for the selection of reference genes). All DNA samples were stored at −20°C. VHL mutation analysis was performed by direct sequencing. Two mutations were detected in the analysed samples (see Table 2). Screening for structural rearrangements, including large deletions was conducted by SB analysis at the University Hospital of Utrecht (The Netherlands). In 14 cases, deletions were identified (see Table 2). A deletion encompassing the entire VHL gene, observed by SB in patients 13 and 16, was confirmed by FISH.
Direct Sequencing
To screen the VHL gene for mutations, we performed direct sequencing of the coding region. Exons 1, 2 and 3 of the VHL gene and their immediately flanking sequences were amplified by PCR, using primers as described by Gnarra et al.18 Note that the oligonucleotide sequence of two primers was published incorrectly, that is, the reverse primer for exon 2 in the downstream intron (5′GTCTATCC TGTACTTACCACAACAACCT3′) and the forward primer for exon 3 in the upstream intron (5′CTGA GACCCTAGTCTGTCACTGAGGAT3′). Instead of using four primer sets for creating partially overlapping fragments, only the forward primer for the first fragment and the reverse primer for the fourth fragment were used for the amplification of exon 1. The PCR amplification products were purified by using the QIAquick PCR Purification Kit (Qiagen), according to the manufacturer's instructions. The amplification primers were used as primers in the sequencing reactions, except for exon 1, for which we designed a new cycle sequencing primer (5′CGAAGATACGGAGGTCGA3′). Cycle sequencing was performed using the ABI PRISM Big Dye Terminator Cycle Sequencing Ready reaction Kit (Applied Biosystems, Foster City, CA, USA), followed by isopropanol precipitation. The fragments were sequenced by automated sequencing analysis on an ABI Prism 377 sequencer (Applied Biosystems).
Southern Blot Analysis
DNA was digested with EcoRI alone, and with an EcoRI/AseI double digest. After gel electrophoresis and transfer to Hybond-N filters, the genomic DNA was hybridised with the VHL g7-cDNA probe, according to the manufacturer's instructions. Upon detection of aberrant fragments, the DNA was digested with at least two other restriction enzymes to exclude the possibility of polymorphisms affecting an EcoRI or AseI site. The human beta globin gene (HBB) was used as an internal control. Additionally, exon-specific probes generated by PCR amplification of exons 1, 2 and 3 were hybridised to the same filters.
Real-Time Quantitative PCR
DNA copy numbers for VHL were determined in an iCycler real-time PCR detection system (Bio-Rad, Hercules, CA, USA) using the DNA-binding dye SYBR Green I. To account for possible variation related to DNA input amounts or the presence of PCR inhibitors, two reference genes ZNF80 and GPR15 were simultaneously quantified in separate tubes for each patient sample.
The method involves amplifications of the three test exons (VHL exons 1, 2 and 3) with unknown copy number and of two reference loci on chromosome 3 with normal copy number (ZNF80 and GPR15). All primers were designed with Primer Express 2.0 software (Applied Biosystems) and tested for specificity using NCBI's BLAST software. Primer sequences are listed in Table 1, and are available in the public RTPrimerDB database19 (http://medgen.ugent.be/rtprimerdb/) (gene (RTPrimerDB-ID): VHL (1023, 1024, 1025), GPR15 (1022) and ZNF80 (1021)). SYBR Green I amplification mixtures (15 μl) contained SYBR Green I master mix (Eurogentec, Seraing, Belgium), 250 nM of each forward and reverse primer, 10 nM fluorescein and 10 ng template DNA.
The cycling conditions were as follows: 10 min at 95°C, 40 cycles at 95°C for 15 s and 60°C for 60 s. After PCR amplification, a melting curve was generated for every PCR product to check the specificity of the PCR reaction (absence of primer–dimers or other nonspecific amplification products). Each assay included: a no-template control (in duplex), 10 ng of calibrator human genomic DNA (Roche Applied Science, Basel, Switzerland, DNA mixture from healthy individuals) (in quadruplicate), 10 ng of SJNB-6 DNA as positive control for deletion (in quadruplicate) and about 10 ng of test DNA (in quadruplicate). The threshold cycle (Ct) values of the iCycler iQ Optical System Software version 3.0 (Bio-Rad) were exported to Excel (Microsoft) for further analysis. Calculation of the gene copy number was performed using the comparative (delta-Ct) Ct method and error propagation rules described in the geNorm manual (http://medgen.ugent.be/~jvdesomp/genorm/).20 The original delta-delta-Ct method21 transforms Ct values into normalised relative target amounts, by relating the Ct value of the target gene in the sample to a calibrator sample and to the Ct value of a reference gene measured in both samples. Note that in the original publication, there is the underlying requirement that the efficiency of target and reference gene should be similar. The delta-Ct method does not employ a reference gene; the Ct value of any gene (either target or reference) is transformed to a quantity relative to a calibrator. This calibrator can be any sample: for example, a real untreated control, or the sample with the highest expression (lowest Ct value). As such, the delta-Ct method generates raw (not yet normalised) quantities, which need to be subsequently normalised by dividing with a proper normalisation factor (the geometric mean of multiple reference genes, as described in Vandesompele et al20). In contrast to the delta-delta-Ct method, the delta-Ct method takes different amplification efficiencies for each gene into account, and allows easy inclusion of multiple reference genes for normalisation, which is a prerequisite for accurate copy number assessment or gene expression analysis. In brief, reaction wells with obvious PCR reaction failure or clear outlier values (difference between Ct and mean Ct greater than 0.3) were excluded from further data analysis. According to the reported standard deviation value of 0.185 on replicated reactions using our real-time PCR machine (http://www.bio-rad.com/LifeScience/pdf/Bulletin_2567.pdf),22 on average one replicate in 10 will be discarded using our criterion. Subsequently, the arithmetic mean of replicated Ct values for each gene is transformed to a relative quantity with the relative quantity of the calibrator set to 1, using the delta-Ct formula  (Q=quantity sample relative to the calibrator sample; E=amplification efficiency (1.97=97%); calibratorCt=Ct value of calibrator), and an amplification efficiency of 97% (determined on the basis of a standard curve consisting of a four-fold DNA dilution series (four points) from normal human genomic DNA (Roche)). For normalisation of the relative quantities, the VHL copy numbers were divided by the geometric mean of two reference gene copy numbers (ZNF80, GPR15). Using this method, a haploid copy number of 1 is expected for a normal sample and a value of 0.5 for a sample with a VHL deletion. For each VHL exon, a value was considered a hemizygous deletion if the haploid copy number was <0.7, and normal when >0.8 (see Results for determination of these cutoff values). A sample with a haploid copy number between these two cutoff values should be reanalysed (not encountered in this study). Errors on replicated Ct values for multiple genes are propagated during Ct transformation to quantities and normalisation using the geometric mean of the two reference genes (ZNF80 and GPR15) according to the geNorm manual and accompanying example calculation file (http://medgen.ugent.be/~jvdesomp/genorm/).20 We developed an Excel template for automated calculations, error propagation and graphical representation of the results (available from the authors on request).
(Q=quantity sample relative to the calibrator sample; E=amplification efficiency (1.97=97%); calibratorCt=Ct value of calibrator), and an amplification efficiency of 97% (determined on the basis of a standard curve consisting of a four-fold DNA dilution series (four points) from normal human genomic DNA (Roche)). For normalisation of the relative quantities, the VHL copy numbers were divided by the geometric mean of two reference gene copy numbers (ZNF80, GPR15). Using this method, a haploid copy number of 1 is expected for a normal sample and a value of 0.5 for a sample with a VHL deletion. For each VHL exon, a value was considered a hemizygous deletion if the haploid copy number was <0.7, and normal when >0.8 (see Results for determination of these cutoff values). A sample with a haploid copy number between these two cutoff values should be reanalysed (not encountered in this study). Errors on replicated Ct values for multiple genes are propagated during Ct transformation to quantities and normalisation using the geometric mean of the two reference genes (ZNF80 and GPR15) according to the geNorm manual and accompanying example calculation file (http://medgen.ugent.be/~jvdesomp/genorm/).20 We developed an Excel template for automated calculations, error propagation and graphical representation of the results (available from the authors on request).
Mfold Analysis
For predicting the secondary structure of single-stranded DNA, we used MFOLD version 3.1 software based on minimal free energy (http://www.bioinfo.rpi.edu/applications/mfold/),23 using default settings and 50 mM Na+, 3 mM Mg2+ and a temperature of 60°C (which is the annealing temperature of the primers).
Results
Assay Design
Selection of reference genes and VHL exon primer pairs
Based on previous experiments, we experienced that accurate discrimination between one and two gene copy numbers is highly dependent on the specific primer pairs used, and the reference genes used for normalisation.15 Appropriate normalisation of quantitative PCR data is required, in order to eliminate nonspecific variation, such as variation in DNA input amounts or presence of PCR inhibitors, especially important in clinical samples. To address this issue, in a first step we evaluated four different candidate reference genes (ZNF80, MOX2, GPR15, HTR1F). Although, in principle any gene can be used as a reference gene, we selected single copy genes that are located on the same chromosome as where the disease gene is located: ZNF80, MOX2 and GPR15 located on the long arm of chromosome 3 (3q13.31, 3q13.2, 3q12.1, respectively), and HTR1F on the short arm, near the centromere (3p12). In order to validate the four reference genes, we determined their copy number in 24 normal DNA samples and analysed the data in the geNorm VBA applet for Microsoft Excel.20 This algorithm was originally designed to determine the most stably expressed reference genes for normalisation of gene expression levels. In this study, it was used to determine the most reliable reference genes in a DNA quantification assay. Based on a gene-stability measure M (which is the average pairwise variation between a particular reference gene and all other tested reference genes), the geNorm program determines the most appropriate genes from a set of tested reference genes in a given sample panel based on the stepwise elimination of the worst-scoring reference gene. Following this approach, it turned out that the differences in stability (ie reproducible and accurate copy number generation) for the four candidate reference genes are very small (M-values ranging between 0.22 and 0.25). Given the fact that we want to use two reference genes for improved normalisation (see further), this means that any combination of two reference genes (from the four evaluated candidate genes) is valuable for normalisation. We selected ZNF80 and GPR15, that is, the two genes with the lowest M-value.
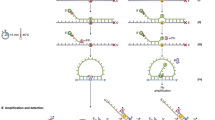
In a subsequent step, we tested three to four different primer pairs for each of the three VHL exons. Test samples included six normal diploid control samples, constitutional DNA from a VHL patient harbouring an entire VHL gene deletion and neuroblastoma cell line SJNB-6 with distal 3p loss (including the VHL locus). Suitable primer pairs were selected upon evaluation of both the melt curve of the amplicon and the measured haploid copy number in the above-mentioned control samples (the theoretical haploid copy number should be 0.5 for the samples harbouring a VHL gene deletion). We noticed that not all primer pairs resulted in accurate quantification (see Figure 1 for different exon 2 primer sets). Inaccurate detection of Ct values for a gene or exon thus appeared to depend on the primer pair used in the amplification process and was shown to be directly related to aberrant PCR amplification efficiency. For the primer pairs that resulted in accurate copy number quantification, we routinely obtained PCR efficiencies higher than 97%; for those that did not, the slope of the standard curve was aberrant (between −1.3 and −2.9), pointing at severe absence of quantitative measurement capacity of these primer pairs due to the secondary structure (absence of primer–dimer or nonspecific amplification was verified for each primer pair). In order to investigate whether the aberrant PCR efficiency and related inaccurate measurement of gene copy number is influenced by the formation of secondary structures in the amplicon, we used MFOLD software based on minimal free energy for predicting the secondary structures of single-stranded DNA.23 If the hybridisation of the primer-target is thermodynamically weaker than the formation of one or more secondary structures, hybridisation of the primer is disfavoured. MFOLD analyses of the different primer sets for exon 2 of the VHL gene are shown in Figure 2. Secondary structures with a melting temperature higher than primer annealing temperature are predicted for amplicons generated by primer sets 2–4. In the case of primer sets 3 and 4, this secondary structure hampers the annealing of the Q-PCR primers, resulting in inconsistent Ct values and inaccurate copy number quantification (see also Figure 1). Similar results were obtained for the tested primer pairs for the other exons, whereby for each exon we selected a suitable primer pair that resulted in smooth and reproducible melting curves, yielding an amplicon without interfering secondary structures. For the reference gene amplicon sequences, no significant secondary structures were found.

Selection of optimal primer pairs for Q-PCR analysis of VHL exon 2. Top: melt curve of four different primer sets for VHL exon 2. Primer pair ‘exon 2 set 2’ demonstrated the most consistent melt curves. Bottom: haploid copy numbers for VHL exon 2, and reference genes GPR15 and ZNF80 in calibrator human genomic DNA (commercial mixture of DNA from healthy individuals), a neuroblastoma cell line with a VHL gene deletion (deletion control) and a normal healthy individual (normal control), with three different primer pairs for exon 2 of the VHL gene. The use of ‘exon 2 set 3’ and ‘exon 2 set 4’ results in incorrect copy number quantification. In contrast, the ‘exon 2 set 2’ provides the expected haploid copy numbers.

Theoretical two-dimensional secondary ssDNA structures of the amplicons generated by four different primer sets for VHL exon 2. Calculated structures in solution of 50 mM Na+ and 3 mM Mg2+ at a temperature of 60°C using version 3.1 MFOLD software. Thermodynamic stability (deltaG in kcal/mole) is presented below the structures. Primers are indicated by the arrows. (a) Amplicon generated by primer set 1: no secondary structure is present at 60°C primers can anneal efficiently; (b) no secondary structure where primer set 2 anneals; the secondary structure between the primer annealing sites has a very small negative deltaG value, and hence does not influence the amplification efficiency; (c) primer set 3 and (d) primer set 4; secondary structure is more stable than primer–target hybridisation; efficient annealing of primers is hampered.
VHL exon copy number quantification
Based on our previously reported Q-PCR strategy for MYCN oncogene copy number determination in neuroblastoma,15 we used SYBR Green I detection chemistry in combination with the comparative Ct method for calculation of the haploid gene copy number. This quantification method is based on differences in Ct value between the test and calibrator sample and avoids the use of standard curves. By using normal control DNA as a calibration sample, a final haploid gene copy number is obtained.
In view of the small difference between the Q-PCR detection values for one and two copies (this is 1 PCR cycle under ideal circumstances), variations in Ct value (and hence variation in copy number) due to pipetting and instrument detection may effect results. To address this issue, we performed four parallel measurements of each sample (ie quadruplicated reactions).
Evaluation of Q-PCR Accuracy, Precision, Sensitivity and Specificity
After optimisation of the Q-PCR method, that is, choice of two reference genes, and selection of optimal primers for quantification of all three VHL exons, a blind VHL deletion screening study was performed on 29 samples for evaluation of sensitivity (false-negative results), specificity (false-positives), accuracy (true-value determination) and precision (random variation) of the Q-PCR approach. The sample test consisted of 17 VHL patients of whom 14 showed partial or entire VHL gene deletion as determined by SB, and 12 normal control samples. Partial or complete deletions were confirmed in 14 of the 17 investigated VHL patients (Table 2) by our quantitative PCR approach. Copy number status was accurately determined in 100% of cases, that is, the Q-PCR and SB results were concordant for all tested samples, thus indicating the outstanding sensitivity and specificity of the Q-PCR approach. For one VHL patient, SB demonstrated a partial deletion of the VHL gene, but was unable to assign the exon that was deleted due to the limited amount of available DNA material. The real-time Q-PCR method revealed a specific deletion of the third VHL exon (Figure 3). To determine the accuracy and to evaluate the precision of the Q-PCR approach, we compared the haploid copy number values and their standard deviations for the deleted vs nondeleted patient samples. No overlap was seen between the two haploid copy numbers in the groups: the mean haploid copy number (±s.d.) being 0.50±0.10 for VHL exon 1 deletion carriers, 0.48±0.08 for VHL patients demonstrating exon 2 deletions, 0.44±0.07 for exon 3-deleted patients (expected value for these three deletions is 0.5) and 1.03±0.08 (exon 1), 0.98±0.08 (exon 2), 1.04±0.11 (exon 3) for normal control samples (expected value is 1.0). Both a nonparametric Mann–Whitney U test and a two-sample t-test (assuming unequal variation) demonstrate that the copy numbers are significantly different between deleted and normal samples (P<10−15). Based on the observed haploid copy number and the standard deviation, we decided to use the mean value of the haploid copy number in deleted samples plus two times the standard deviation as upper limit for scoring a deletion, and mean value of the normal haploid copy number minus two times the standard deviation as lower limit. As such, samples with a haploid copy number ≤0.7 were scored as deleted, while a haploid copy number ≥0.8 points at a normal copy number status (Figure 4).

VHL copy number status (±standard error of the mean) determined by Q-PCR. Haploid copy number for the three VHL gene exons and the reference genes ZNF80 and GPR15 in normal genomic DNA (calibrator), neuroblastoma cell line (SJNB-6) demonstrating distal 3p loss (including the VHL locus) and two test samples: sample 13 (entire gene deletion) and sample 14 (VHL exon 3 deletion). Calculations and graphical representation were performed using our Excel template (see Materials and methods).

Haploid copy number values for VHL deletion patients and normal controls. Box-plot representation of haploid copy numbers obtained for VHL patients harbouring an exon or entire gene deletion (14), the deletion control SJNB-6 cell line (17 independent assays) and normal controls (12) (median, box: 25–75th percentile, whiskers: lines that extend from the box to the highest and lowest values, excluding outliers; circle: outlier). The upper and lower cutoff levels of 0.7 and 0.8 for scoring a deletion or normal copy number status, respectively, are indicated.
Subsequently, we tested whether two reference genes (instead of one) are needed for proper normalisation. The use of only one reference gene (ZNF80 or GPR15) resulted in an overlap between the haploid copy numbers calculated for the deleted and normal samples (data not shown), meaning that a haploid copy number value of a deleted sample occasionally was found to be higher than one for a normal status. Hence, no general cutoff values for deleted vs nondeleted samples could be determined using only one reference gene. Furthermore, standard deviations on calculated copy numbers were systematically higher (up to 0.07 U) when only one reference gene was used for normalisation instead of two. These findings clearly demonstrate the improved accuracy and precision (lower standard deviation) obtained by using two reference genes.
Discussion
The VHL syndrome (OMIM 199300) is a dominantly inherited cancer syndrome predisposing to a variety of malignant and benign neoplasms that include retinal haemangioblastomas, haemangioblastomas of the central nervous system, renal cell carcinomas, pancreatic cysts and tumours, phaeochromocytomas, endolymphatic sac tumours and epididymal cystadenomas.3, 16, 17 VHL is caused by germline mutations of one allele of the VHL tumour suppressor gene, followed by somatic inactivation or loss of the remaining wild-type allele. Germline mutations have been detected in up to 100% of VHL kindred.4 Mis-sense, nonsense, splice-site mutations, base pair deletions and insertions have been detected in approximately 80% of these families. In the remaining 20% of the VHL families, exon or entire gene deletions were observed.3, 4, 5
In this study, we report the design and application of a real-time Q-PCR test for the detection of exon or entire gene deletions of the VHL gene. For many years, SB analysis and FISH were the golden standard for this type of analysis. However, these methods have several intrinsic limitations. SB is laborious, time-consuming, often uses radioactive isotopes and requires large amounts of high-quality DNA. FISH allows the visualisation of large chromosomal deletions at the single-cell level, but is not well suited for detection of partial gene deletions. In contrast, real-time Q-PCR has evolved as a valuable alternative in molecular diagnostics. This method has many advantages, including the absence of post-PCR manipulation (significantly reducing contamination risk and hands-on time), its speed, sensitivity and large dynamic range of accurate quantification. All these factors make this technology very well suited for routine diagnosis. Hence, real-time PCR machines have found their way in diagnostic labs, among others for pathogen detection, and quantification of fusion genes in malignancies.
Here, we describe a real-time Q-PCR approach for the detection of single, multiexon or entire gene deletions. We have clearly shown that for accurate quantification and discrimination between one vs two gene copies, primer design and normalisation using two reference genes instead of one proved to be critical factors. Amplicons should be free of secondary structures, which hamper efficient primer annealing and extension, and hence disturb accurate quantification, as demonstrated for some primer pairs designed for exon 2 of the VHL gene. In our search for proper reference genes, we experienced that all tested primer pairs yielding secondary structure-free amplicons are equally suited for normalisation. As it turned out, the geNorm algorithm (originally developed for identification of stably expressed reference or the so-called housekeeping genes) pointed at only minor differences in ‘stability’ (ie reproducible and accurate copy number generation) for the four tested candidate reference genes. After selection and quantification of two reference genes, we demonstrated that the use of two reference loci instead of one single locus for normalisation of the data not only allowed internal quality control but also resulted in improved accuracy and precision.
To accommodate the numerous calculations involved in transforming Ct values to normalised haploid copy numbers, we also developed an easy-to-use and freely available Excel template for automated data analysis and graphical representation.
For evaluation of our optimised Q-PCR assay, we tested in a blind study whether all partial/complete gene deletions could be detected, and determined in parallel the diagnostic cutoff values for a deleted or normal copy number status, using the Ct values of 29 samples with a known genotype (15 with normal gene copy number status, 14 exon/gene deletions). The upper and lower cutoff values for deletion and normal haploid copy numbers, respectively, were 0.7 and 0.8, based on mean haploid copy number value±two times the standard deviation, demarcating a safety margin of 0.1 (Figure 4). This large margin, mainly attributed to the use of two reference genes, points at accurate and precise copy number measurements. As mentioned above, the use of only one reference gene provides less accurate results, as there is no longer a safety margin. The blind deletion screening study resulted in correct classification of all samples according to their gene copy number status, illustrating the high sensitivity and specificity of the Q-PCR assay for detecting germline deletions in the VHL gene.
Recently, two other PCR-based methods have been described for the detection of gene copy number changes: multiplex-amplifiable probe hybridisation13 and MLPA.14 Both techniques rely on a combination of hybridisation of multiple probes to genomic DNA, subsequent PCR amplification of the probes and (capillary) gel electrophoresis. These methods have been applied for the detection of duplications and deletions whereby the power of these techniques lies in the simultaneous analysis of multiple sites in the genome. Choice between MLPA and Q-PCR in part depends on the availability of a real-time PCR machine or (capillary) sequencer/electrophoresis system. A major advantage of our Q-PCR assay is the speed by which an analysis can be performed. Using the described assay for VHL exon deletion screening, two unknown patient samples and a positive and negative control for deletion can be analysed in one 96-well reaction plate in 3.5 h, including 1 h hands-on time and data analysis using our Excel template. MLPA is far more laborious and time-consuming since the MLPA DNA quantification protocol consists of multiple steps, that is, DNA denaturation and hybridisation of the probes (incubation at 60°C for 16 h), a ligation reaction, PCR amplification, separation of the amplification products and data analysis. For Q-PCR, only limited amounts of input DNA are required (10 ng of input DNA per PCR reaction; 200 ng for the complete assay). Although it was reported that 20 ng of DNA is sufficient to work with for MLPA analysis, Sellner and Taylor24 found that 100–200 ng is required for reliable and reproducible results. Reagent costs for Q-PCR and MLPA are comparable, although MLPA is more cost-effective for the simultaneous analysis of multiple (>5) samples. It is important to note, however, that for a relatively rare disorder, only few patients need to be tested at the same time. Hence, in view of the limited shelf life of MLPA kits, Q-PCR might turn out to be a more flexible and economical alternative.
In conclusion, we outlined a technical framework and provided general assay guidelines for PCR-based gene copy number quantifications. We applied our quantification strategy on the VHL gene, and showed that real-time quantitative PCR allows sensitive, specific, precise and accurate detection of VHL germline deletions. Therefore, this approach should be implemented as the method of choice in routine DNA diagnosis for VHL disease. The presented approach is easily adaptable for the screening of DNA copy number alterations in other genetic diseases.
References
Herman JG, Latif F, Weng Y, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA 1994;91:9700–9704.
Prowse AH, Webster AR, Richards FM, et al. Somatic inactivation of the VHL gene in von Hippel–Lindau disease tumors. Am J Hum Genet 1997;60:765–771.
Maher ER, Webster AR, Richards FM, et al. Phenotypic expression in von Hippel–Lindau disease: correlations with germline VHL gene mutations. J Med Genet 1996; 33:328–332.
Stolle C, Glenn G, Zbar B, et al. Improved detection of germline mutations in the von Hippel–Lindau disease tumor suppressor gene. Hum Mutat 1998;12:417–423.
Zbar B, Kishida T, Chen F, et al. Germline mutations in the von Hippel–Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat 1996;8:348–357.
Stenson PD, Ball EV, Mort M, et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat 2003;21:577–581.
Yao M, Latif F, Orcutt ML, et al. von Hippel–Lindau disease: identification of deletion mutations by pulsed-field gel electrophoresis. Hum Genet 1993;92:605–614.
Richards FM, Phipps ME, Latif F, et al. Mapping the von Hippel–Lindau disease tumour suppressor gene: identification of germline deletions by pulsed field gel electrophoresis. Hum Mol Genet 1993;2:879–882.
Decker HJ, Klauck SM, Lawrence JB, et al. Cytogenetic and fluorescence in situ hybridization studies on sporadic and hereditary tumors associated with von Hippel–Lindau syndrome (VHL). Cancer Genet Cytogenet 1994;77:1–13.
Pack SD, Zbar B, Pak E, et al. Constitutional von Hippel–Lindau (VHL) gene deletions detected in VHL families by fluorescence in situ hybridization. Cancer Res 1999;59:5560–5564.
Phipps ME, Maher ER, Affara NA, et al. Physical mapping of chromosome 3p25–p26 by fluorescence in situ hybridisation (FISH). Hum Genet 1993;92:18–22.
Cybulski C, Krzystolik K, Maher ER, et al. Long polymerase chain reaction in detection of germline deletions in the von Hippel–Lindau tumour suppressor gene. Hum Genet 1999;105:333–336.
Armour JA, Sismani C, Patsalis PC, et al. Measurement of locus copy number by hybridisation with amplifiable probes. Nucl Acids Res 2000;28:605–609.
Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucl Acids Res 2002;30:e57.
De Preter K, Speleman F, Combaret V, et al. Quantification of MYCN, DDX1, and NAG gene copy number in neuroblastoma using a real-time quantitative PCR assay. Mod Pathol 2002;15:159–166.
Maher ER, Yates JR, Harries R, et al. Clinical features and natural history of von Hippel–Lindau disease. Q J Med 1990;77:1151–1163.
Maher ER, Kaelin Jr WG . von Hippel–Lindau disease. Medicine (Baltimore) 1997;76:381–391.
Gnarra JR, Duan DR, Weng Y, et al. Molecular cloning of the von Hippel–Lindau tumor suppressor gene and its role in renal carcinoma. Biochim Biophys Acta 1996;1242:201–210.
Pattyn F, Speleman F, De Paepe A, et al. RTPrimerDB: the real-time PCR primer and probe database. Nucl Acids Res 2003;31:122–123.
Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:RESEARCH0034.
Livak KJ, Schmittgen TD . Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001;25: 402–408.
Boeckman F, Hamby K, Tan L . Real-time PCR using the iCycler iQ detection system and intercalating dyes. Tech note 2620.
Zuker M . Mfold web server for nucleic acid folding and hybridization prediction. Nucl Acids Res 2003;31: 3406–3415.
Sellner LN, Taylor GR . MLPA and MAPH: new techniques for detection of gene deletions. Hum Mutat 2004;23:413–419.
Acknowledgements
Jo Vandesompele is supported by a grant from the Flemish Institute for the Promotion of Innovation by Science and Technology in Flanders (IWT). This study is supported by GOA-Grant 12051397, FWO-Grant G.0185.04 and VEO Project 011V1302. This text presents research results of the Belgian program of Interuniversity Poles of attraction initiated by the Belgian State, Prime Minister's Office, Science Policy Programming (IUAP).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hoebeeck, J., van der Luijt, R., Poppe, B. et al. Rapid detection of VHL exon deletions using real-time quantitative PCR. Lab Invest 85, 24–33 (2005). https://doi.org/10.1038/labinvest.3700209
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700209
Keywords
This article is cited by
-
Evaluation of the functional effects of genetic variants‒missense and nonsense SNPs, indels and copy number variations‒in the gene encoding human deoxyribonuclease I potentially implicated in autoimmunity
Scientific Reports (2019)
-
The contribution of 7q33 copy number variations for intellectual disability
neurogenetics (2018)
-
Inhibition of neuronal FLT3 receptor tyrosine kinase alleviates peripheral neuropathic pain in mice
Nature Communications (2018)
-
Impairment of IGF2 gene expression in prostate cancer is triggered by epigenetic dysregulation of IGF2-DMR0 and its interaction with KLF4
Cell Communication and Signaling (2017)
-
High-throughput PCR assay design for targeted resequencing using primerXL
BMC Bioinformatics (2017)
