
Abstract
Ovarian serous cystadenomas are common ovarian lesions that may be precursors of serous borderline tumors, which can in turn progress to low-grade serous carcinomas. It has been shown that low-grade serous carcinoma and serous borderline tumors are characterized by frequent mutations in BRAF or KRAS genes, but the mutational status of these genes in serous cystadenomas and the clonal nature of serous cystadenomas have not been fully investigated. We isolated cyst-lining epithelium from 30 consecutive serous cystadenomas, and analyzed their BRAF and KRAS mutational status. Wild-type sequences of BRAF and KRAS were detected in all specimens. Using the human androgen receptor gene as a polymorphic marker, we also examined the clonal status of epithelial cells in all of the serous cystadenomas. Four of 29 (14%) informative specimens were monoclonal based on the methylation pattern. These monoclonal cystadenomas were significantly (P<0.01) larger in size (>8 cm) than the nonclonal cystadenomas. These data indicate that serous cystadenomas do not contain mutations in either BRAF or KRAS genes and that most serous cystadenomas are polyclonal. Accordingly, it appears that serous cystadenomas develop as a hyperplastic expansion from epithelial inclusions with a clonal/neoplastic transformation occurring in a subset of them.
Similar content being viewed by others

Main
Ovarian carcinoma is believed to arise either from the surface epithelium covering the ovary, benign epithelial inclusions or cystic tumors termed ‘cystadenomas’. The World Health Organization classification divides ovarian surface epithelial tumors into three groups: benign cystadenomas, invasive carcinomas and an intermediate group designated ‘borderline’ tumors.1, 2 Ovarian cystadenomas are further subclassified according to the cell type into serous cystadenomas, mucinous cystadenomas and endometriomas.3, 4, 5, 6, 7 Cystadenomas occur in 5–15% of postmenopausal women in the general population.8, 9 Serous cystadenomas are the most common followed by mucinous cystadenomas and endometriomas.10 Previous studies have demonstrated that mucinous cystadenomas11, 12 and endometriomas13 are clonal and therefore are benign neoplasms, but similar studies of serous cystadenomas have not been performed to our knowledge.
Based on our previous morphological and molecular studies, we have proposed that serous borderline tumors (atypical proliferative tumors and noninvasive micropapillary serous carcinomas) can progress to low-grade serous carcinomas (invasive micropapillary serous carcinomas).14, 15 These low-grade serous carcinomas and borderline tumors exhibit frequent BRAF or KRAS mutations but rare p53 mutations.6, 14, 16 In contrast, high-grade serous carcinomas have wild-type KRAS and BRAF but frequent p53 mutations even when they are small and confined to the ovary.17, 18 It has therefore been proposed that high-grade serous carcinomas in contrast to low-grade (invasive micropapillary) serous carcinomas develop directly from epithelial inclusions or ovarian surface epithelium, so-called, ‘de novo’ development.14, 19 In order to elucidate the mechanisms of serous carcinogenesis, specifically the molecular genetic alterations in early tumorigenesis, we undertook a study of serous cystadenomas aimed at assessing whether mutations of BRAF and KRAS, which are common in serous borderline tumors and low-grade serous carcinomas are present in serous cystadenomas.6, 19, 20, 21 In addition, we determined the clonality of serous cystadenomas by analyzing the patterns of X-chromosome inactivation of the X-linked androgen receptor (HUMARA) gene. For molecular genetic analyses, we have applied a new method to isolate pure and abundant cyst-lining epithelial cells from fresh serous cystadenomas. Finally, we compared the proliferative and apoptotic activity of the epithelium of the cystadenomas to that of ovarian surface epithelium and serous borderline tumors.
Materials and methods
Epithelial Cell Isolation from Serous Cystadenomas
A total of 30 consecutive ovarian serous cystadenomas were collected from women undergoing oophorectomy at the Johns Hopkins Hospital, Baltimore, Maryland. One serous borderline tumor was also included as a control. The study protocol was approved by the local institutional review board. Frozen sections of the cystadenomas were performed to confirm the diagnosis before harvesting the epithelial cells from at least half of the sample. For multilocular cystadenomas, the largest one was used in the assays. In order to obtain abundant and pure cyst-lining epithelial cells for multiple molecular genetic analyses, we employed a method to enrich the epithelial cells by combining chemical or mechanical separation and immunosorting. After washing in phosphate buffer saline (PBS) 3 times, the cyst-lining was incubated in 0.05% trypsin and 0.53 mM EDTA (Invitrogen, Grand Island, NY, USA) at 37°C for 10 min, then mechanically separated from the cyst wall by agitating the cyst fragments and/or gentle scraping using a rubber cell scraper (Sarstedt, Newton, NC, USA). The epithelial cell fragments were collected and washed in PBS (Figure 1). The epithelial cells were enriched by Epi-CAM-conjugated Dynal Beads (Dynal, Hamburg, Germany) that specifically bound epithelial cells. The procedures were detailed in the vendor's instructions. The purity of isolated epithelial cells was assessed by immunostaining for the expression of cytokeratin 8 (an epithelial marker), using the antibody CAM5.2 (Becton Dickinson, San Jose, CA)22 on the isolated cells after short-term culture in 24-well plates. The isotype-matched MN-4 antibody that reacted with Mel-CAM (CD146) was also used as a negative control.23 The immunofluorescence staining and nuclear counterstaining were detailed in the previous report.24

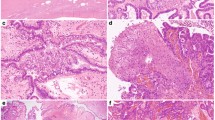
Isolation of cyst-lining epithelium. (a) The lining epithelium was gently scraped from the cyst wall to the right of the arrow; the epithelium to the left of the arrow was undisturbed (hematoxylin and eosin stain). (b): Immunostains for collagen IV demonstrate the basement membrane. The epithelium is present to the left of the arrow and absent to the right of the arrow that has been harvested. (c) Immunostaining for CK8 antibody shows green fluorescence in the cytoplasm of isolated epithelial cells in all the cells. Nuclei are counterstained red. (d) Immunostaining for CD146 antibody as a negative control. There is no green fluorescence present.
Mutational Analysis
Nucleotide sequencing was used to analyze the mutational status of BRAF and KRAS. The primers that were used to amplify exon 15 of BRAF and exon 1 of KRAS containing codons 12 and 13 and the PCR protocols have been previously described.14, 25, 26 PCR products were purified using a MiniElute™ PCR purification kit (Qiagen, Valencia, CA, USA). Nucleotide sequencing was performed using fluorescent labeled Applied Biosystems Big Dye terminators and an Applied Biosystems 377 automated sequencer (Applied Biosystems, Foster City, CA, USA). Analysis of the 1796T/A status in BRAF was also performed using a PCR-based restriction fragment length polymorphism (RFLP) technique as previously described.27, 28 For this method, the BRAF PCR product of exon 15 containing the relevant nucleotide at position 1796 was digested with TspR1 (New England BioLabs, Beverly, MA, USA) at 65°C for 3 h. The samples were electrophoresed on a 10% polyacrylamide gel.
Clonality Assay
The clonal status of the cystadenomas was evaluated by analyzing the patterns of X-chromosome inactivation in exon 1 of the X-linked androgen receptor gene (HUMARA), which contains a highly polymorphic trinucleotide repeat.29, 30 The QiaQuick PCR purification kit (Qiagen, Valencia, CA, USA) was used to isolate genomic DNA from the purified cyst epithelial cells from all the 30 specimens. In addition, a serous borderline tumor served as a positive control. Predigestion of DNA (2 μg) with 20 U of methylation-sensitive restriction endonuclease HhaI or HpaII (New England BioLabs, Beverly, MA, USA) resulted in selective PCR amplification from the methylated (uncleaved) allele. For PCR, 3 μl of the enzyme digest was mixed with primers and all essential PCR reagents as previously reported.30 The PCR product was separated in 6% polyacrylamide gels and visualized by ethidium bromide.
Assessment of Proliferation and Apoptotic Activity
The proliferative and apoptotic activity of the cyst-lining epithelial cells was assessed by immunohistochemistry using the MIB1 antibody to detect Ki-6731, 32 and the M30 antibody33 to detect the cytokeratin epitope after apoptosis, respectively. The immunohistochemistry methodology has been previously described.34 Paraffin sections from 10 normal ovaries, 50 serous cystadenomas and 15 serous borderline tumors were stained with the MIB1 and the M30 antibody. At least 4000 epithelial cells were randomly selected from different regions and analyzed using the Spotlight morphometric program under a Nikon inverted light microscope (Image System, Columbia, MD, USA). The Ki-67 and M30 labeling index was expressed as the percentage of Ki-67 or M30 labeled epithelial cells from among the total number of epithelial cells counted. Two-sided Student's t-test was used to compare the difference in the Ki-67 and M30 labeling index between the serous cystadenomas, normal surface ovarian epithelium and serous borderline tumors.
Results
The 30 serous cystadenomas ranged from 1.2 to 10 cm in greatest dimension with a median of 5 cm (Table 1). Of the 30 cystadenomas, 26 were unilocular and the remaining four cystadenomas were multilocular (specimen no. 15, 17, 21 and 29). The epithelial cells lining the cystadenomas ranged from flattened to columnar (Figure 1). The latter cells frequently contained cilia (serous differentiation) at least in focal areas. The nuclei were bland and none displayed cytologic atypia (so-called dysplasia). Immunostaining revealed that more than 98% of the cells isolated from cystadenomas were positive for cytokeratin 8, confirming the purity of the epithelial cells isolated by our technique (Figure 1). Mutational analysis with direct nucleotide sequencing demonstrated that none of the samples harbored mutations in either BRAF (exon 15) or KRAS (exon 1) gene. As a control, the epithelial cells isolated from a serous borderline tumor revealed a missense mutation at the codon 12 of KRAS (GGT to GAT). The presence of wild-type BRAF gene in all of the serous cystadenomas was confirmed using a restriction fragment length polymorphism assay that detects rare mutations at codon 599 of BRAF.27, 28
The lack of mutations in BRAF and KRAS raises the possibility that serous cystadenomas may not be clonal. Therefore, we determined the clonality of all serous cystadenomas by analyzing patterns of X-chromosome inactivation of the X-linked androgen receptor (HUMARA). All the samples except one were heterozygous and therefore informative for clonal analysis (Table 1). The positive control specimen, a serous borderline tumor, demonstrated a monoclonal composition, as one of the alleles was absent after enzyme digestion. Based on HUMARA assay, we found that only four of the 29 (14%) of the serous cystadenomas showed a clonal pattern (Figure 2 and Table 1). The clonal serous cystadenomas tended to be larger in size (>8 cm) than the polyclonal serous cystadenomas (P<0.01; two-tailed t-test).

Clonality assay with the polymorphic markers of the HUMARA gene. Representative cystadenoma samples (25 and 26) are shown. Sample 25 retains all the bands in HpaII (E1) and HhaI (E2) predigested samples as compared to the control with buffer only (B). In contrast, sample 26 shows an absence of the shorter band (arrow) in enzyme-digested samples, indicating a homogenous methylation pattern, that is, monoclonality, in this sample.
The Ki-67 labeled epithelial cells were scattered randomly, not clustered, as single positive cells in the serous cystadenoma with a frequency of 0.84±0.33%. This proliferative index was significantly higher than the 0.10±0.028% in normal ovarian surface epithelium (P<0.001) and significantly lower than the 8.15±2.80% in serous borderline tumors (P<0.001) (Figure 3). The apoptotic index as determined by the M30 immunoreactivity was extremely low (<0.01%) in serous cystadenomas, as well as in ovarian surface epithelium and serous borderline tumors. There was no statistically significant difference in Ki-67 or M30 labeling index (P>0.1) between clonal and polyclonal cystadenomas nor did the size of the cystadenomas correlate with the Ki-67 or M30 labeling index (data not shown).

Proliferative activity in ovarian surface epithelium (OSE), serous cystadenomas and serous borderline tumors (SBT) as determined by the Ki-67 labeling index. Serous cystadenomas have a significantly higher Ki-67 labeling index than ovarian surface epithelium (P<0.001) but a lower index than serous borderline tumors (P<0.001).
The time for a serous cystadenomas to double in diameter was calculated by assuming that the cystadenomas are spheres with a diameter of A cm and that the epithelium evenly lined the inner surface of the cyst. The total number of epithelial cells in a cyst (NA) was equal to the total inner surface area of a cyst (A2π)/the (en face) area of an individual epithelial cell which was 12 × 12 μm based on morphometric measurements. For a cyst with a diameter of A cm to enlarge to a diameter of 2A cm, a four fold increase in cell number is required since N2A/NA=4A2π/144 μm2/A2π/144 μm2=4. Given the net proliferation index (Ki-67 index−M30 index) of 0.84% and a presumable cell doubling time of t hours in cyst-lining epithelial cells, it was estimated that it would take 200/0.84 × t hours for a cyst with an original cell number of NA to increase to 4 NA (N2A). The cell doubling time, t, depends on the cell type and the microenvironment, and generally is between 48 and 96 h. Therefore, given t=72 h, the estimated time for a serous cyst to double in size was 200/0.84 × 72 h=17.143 h=714 days or 23.8 months.
Discussion
Clonality and mutations are the molecular signatures of the vast majority of neoplasms.35 Accordingly, our data demonstrating an absence of BRAF and KRAS mutations in all of the serous cystadenomas in this analysis and an absence of clonality in the vast majority of them provide cogent evidence that most so-called ‘serous cystadenomas’ are not neoplasms. This conclusion is supported by other studies showing a diploid chromosomal content36 and lack of p53 mutations in serous cystadenomas.6 The Ki-67 immunohistochemical findings in the present study indicating that the epithelial cells of these cystadenomas do proliferate, albeit at a very low rate, suggest that the development of a nonclonal serous cyst is a hyperplastic process. The mechanism underlying increased proliferative activity in the cyst-lining epithelium as compared to the ovarian surface epithelium is unknown, but it may be related to the sustained hydrostatic pressure that has been shown to induce cellular proliferation.37, 38, 39 The mildly increased proliferation index could explain why serous cystadenomas can reach a size as large as 10 cm. Based on our estimates, the time for a cyst to double in size (diameter) is approximately 2 years. Therefore, it may take 16 years for a small inclusion cyst of 300 μm in diameter to enlarge to a 10 cm cyst. It should be noted that the doubling time of a cyst is based on estimation and in fact it depends on several factors, especially the cell cycle transit time of proliferating cyst-lining epithelium, which is difficult to measure in vivo. It is apparent that not all serous inclusion cysts will progress to large cysts and further studies are required to investigate the growth kinetics of cysts and molecular mechanisms underlying their development. The present study demonstrates that ovarian serous cystadenomas do not contain mutations in either BRAF or KRAS genes and that most are polyclonal. In previous studies, we reported that low-grade serous carcinomas develop in a stepwise fashion from serous borderline tumors.14, 15 Although the molecular events that lead to the development of serous borderline tumors are not known, our findings suggest that activating mutations in BRAF or KRAS may play an important role as both mutations are found in over 60% of serous borderline tumors and in low-grade serous carcinomas.27 This view is supported by the observation that activating mutations in these genes are oncogenic in experimental cell culture systems.25, 40, 41 Accordingly, the clonal nature of only a small proportion of serous cystadenomas and the absence of KRAS or BRAF mutations in them suggests that mutations in these genes may be a key early event in the transformation of clonal serous cystadenomas to serous borderline tumors.
Although the findings in this report indicate that 86% of ovarian serous cystadenomas are polyclonal and non-neoplastic, this may be an overestimate as monoclonality may have been undetected in some cases for several reasons. First, for the genetic analysis, we pooled the bulk of the cyst-lining epithelium from each specimen to avoid the bias introduced by the X-inactivation patch size in the assessment of clonality.42 It is possible that clonal proliferation occurs only focally in some cystadenomas and that the molecular changes in these foci occur in the absence of morphologic alterations that are not detected microscopically. Second, our results do not exclude the possibility that multiple independent clonal events occur in a cyst, which would then result in the erroneous impression of polyclonality. Third, since the monoclonal cystadenomas tended to be large and the number of large cystadenomas (>8 cm) that were analyzed in this study was relatively small, it is possible that the proportion of monoclonal serous cystadenomas would have been higher if a greater number of large cystadenomas were analyzed. Finally, very small inclusion cysts (<1 cm) were not analyzed in this study because they are too small and by definition do not qualify as cystadenomas.43
In conclusion, the data in this report demonstrate that mutations in BRAF and KRAS that characterize serous borderline tumors and low-grade serous carcinomas are absent in serous cystadenomas. In fact, only 14% of serous cystadenomas are clonal, suggesting that serous cystadenomas develop as a hyperplastic expansion of ovarian surface epithelial inclusions. We speculate that a small proportion of these cystadenomas become clonal and that mutations of KRAS or BRAF in some of these clonal cystadenomas lead to the development of serous borderline tumors, which are the precursors of low-grade serous carcinoma. These findings have important implications for understanding the pathogenesis of ovarian serous carcinoma, and for the screening and treatment of ovarian cancer.
References
Serov SF, Scully RE, Sobin LH . International Hisotlogical Classification and Staging of Tumors, Histologic Typing of Ovarian Tumors. World Health Organization: Geneva, 1973.
Scully RE . World Health Organization International Histological Classification of Tumours, 2 edn. Springer: New York, NY, 1999.
Feeley KM, Wells M . Precursor lesions of ovarian epithelial malignancy. Histopathology 2001;38:87–95.
Scully RE . Pathology of ovarian cancer precursors. J Cell Biochem 1995;23(Suppl):208–218.
Scully RE . Early de novo ovarian cancer and cancer developing in benign ovarian lesions. Int J Gynaecol Obstet 1995;49(Suppl):S9–S15.
Zheng J, Benedict WF, Xu HJ, et al. Genetic disparity between morphologically benign cysts contiguous to ovarian carcinomas and solitary cystadenomas [see comments]. J Natl Cancer Inst 1995;87:1146–1153.
Hutson R, Ramsdale J, Wells M . p53 protein expression in putative precursor lesions of epithelial ovarian cancer. Histopathology 1995;27:367–371.
Oyelese Y, Kueck AS, Barter JF, et al. Asymptomatic postmenopausal simple ovarian cyst. Obstet Gynecol Surv 2002;57:803–809.
Christensen JT, Boldsen JL, Westergaard JG . Functional ovarian cysts in premenopausal and gynecologically healthy women. Contraception 2002;66:153–157.
Seidman JD, Russell P, Kurman RJ . Surface epithelial tumors of the ovary. In: Kurman RJ (ed). Blaustein's Pathology of the Female Genital Tract, 5th edn. Springer-Verlag: New York, 2002, pp 791–904.
van der Bijl AE, Fleuren GJ, Kenter GG, et al. Unique combination of an ovarian gonadoblastoma, dysgerminoma, and mucinous cystadenoma in a patient with Turner's syndrome: a cytogenetic and molecular analysis. Int J Gynecol Pathol 1994;13:267–272.
Cuatrecasas M, Villanueva A, Matias-Guiu X, et al. K-ras mutations in mucinous ovarian tumors: a clinicopathologic and molecular study of 95 cases. Cancer 1997;79:1581–1586.
Jimbo H, Hitomi Y, Yoshikawa H, et al. Evidence for monoclonal expansion of epithelial cells in ovarian endometrial cysts. Am J Pathol 1997;150:1173–1178.
Singer G, Kurman RJ, Chang H-W, et al. Diverse tumorigenic pathways in ovarian serous carcinoma. Am J Pathol 2002;160:1223–1228.
Sehdev AES, Sehdev PS, Kurman RJ . Noninvasive and invasive micropapillary serous carcinoma of the ovary: a clinicopathologic analysis of 135 cases. Am J Surg Pathol 2003;27:725–736.
Deligdisch L . Ovarian dysplasia: a review. Int J Gynecol Cancer 1997;7:89–94.
Shelling AN, Cooke I, Ganesan TS . The genetic analysis of ovarian cancer. Br J Cancer 1995;72:521–527.
Pothuri B, Leitao M, Barakat R, et al. Genetic analysis of ovarian carcinoma histogenesis. Gynecol Oncol 2001;80:277.
Teneriello MG, Ebina M, Linnoila RI, et al. p53 and Ki-ras gene mutations in epithelial ovarian neoplasms. Cancer Res 1993;53:3103–3108.
Cuatrecasas M, Erill N, Musulen E, et al. K-ras mutations in nonmucinous ovarian epithelial tumors: a molecular analysis and clinicopathologic study of 144 patients. Cancer 1998;82:1088–1095.
Kappes S, Milde-Langosch K, Kressin P, et al. p53 mutations in ovarian tumors, detected by temperature-gradient gel electrophoresis, direct sequencing and immunohistochemistry. Int J Cancer 1995;64:52–59.
Moll R, Franke WW, Schiller DL, et al. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell 1982;31:11–24.
Shih IM, Nesbit M, Herlyn M, et al. A new Mel-CAM (CD146)-specific monoclonal antibody, MN-4, on paraffin-embedded tissue. Mod Pathol 1998;11:1098–1106.
Shih IM, Yu J, He TC, et al. The beta-catenin binding domain of adenomatous polyposis coli is sufficient for tumor suppression. Cancer Res 2000;60:1671–1676.
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–954.
Vogelstein B, Kinzler KW . Digital PCR. Proc Natl Acad Sci USA 1999;96:9236–9241.
Singer G, Oldt III R, Cohen Y, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst 2003;95:484–486.
Cohen Y, Zhao XM, Mambo E, et al. BRAF mutation occurs in a majority of papillary thyroid carcinoma. J Natl Cancer Inst 2003;95:625–627.
Enomoto T, Fujita M, Inoue M, et al. Analysis of clonality by amplification of short tandem repeats. Carcinomas of the female reproductive tract. Diagn Mol Pathol 1994;3:292–297.
Allen RC, Zoghbi HY, Moseley AB, et al. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 1992;51:1229–1239.
Gerdes J, Schwab U, Lemke H, et al. Production of a mouse monoclonal antibody reactive with a human nuclear antigen associated with cell proliferation. Int J Cancer 1983;31:13–20.
Gerdes J, Lemke H, Baisch H, et al. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 1984;133:1710–1715.
Leers M, Kolgen W, Bjorklund V, et al. Immunocytochemical detection and localization of a cytokeratin 18 epitope exposed during early apoptosis. J Pathol 1999;5:567–572.
Shih IM, Kurman RJ . Ki-67 labeling index in the differential diagnosis of exaggerated placental site, placental site trophoblastic tumor, and choriocarcinoma: a double immunohistochemical staining technique using Ki-67 and Mel-CAM antibodies. Hum Pathol 1998;29:27–33.
Vogelstein B, Kinzler KW . The Genetic Basis of Human Cancer. McGraw-Hill Health Professions Division: New York, 1998.
Kotylo PK, Michael H, Fineberg N, et al. Flow cytometric analysis of DNA content and RAS P21 oncoprotein expression in ovarian neoplasms. Int J Gynecol Pathol 1992;11:30–37.
Acevedo AD, Bowser SS, Gerritsen ME, et al. Morphological and proliferative responses of endothelial cells to hydrostatic pressure: role of fibroblast growth factor. J Cell Physiol 1993;157:603–614.
Haberstroh KM, Kaefer M, Retik AB, et al. The effects of sustained hydrostatic pressure on select bladder smooth muscle cell functions. J Urol 1999;162:2114–2118.
Hirokawa M, Miura S, Kishikawa H, et al. Loading of mechanical pressure activates mitogen-activated protein kinase and early immediate gene in intestinal epithelial cells. Dig Dis Sci 2001;46:1993–2003.
Peyssonnaux C, Eychene A . The Raf/MEK/ERK pathway: new concepts of activation. Biol Cell 2001;93:53–62.
Malumbres M, Barbacid M . RAS oncogenes: the first 30 years. Nat Rev Cancer 2003;3:459–465.
Novelli M, Cossu A, Oukrif D, et al. X-inactivation patch size in human female tissue confounds the assessment of tumor clonality. Proc Natl Acad Sci USA 2003;27:27.
Scully RE, Young RH, Clement PB . Surface epithelial-stromal tumours and serous tumors. In: Rosai J (ed). Tumors of the Ovary, Maldeveloped Gonads, Fallopian Tube, and Broad Ligament, Third series edn. Armed Forces Institute of Pathology: Washington, DC, 1998, pp 51–79.
Acknowledgements
This work was supported by the research Grant OC010017 from the US Department of Defense.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cheng, E., Kurman, R., Wang, M. et al. Molecular genetic analysis of ovarian serous cystadenomas. Lab Invest 84, 778–784 (2004). https://doi.org/10.1038/labinvest.3700103
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3700103
Keywords
Keywords
This article is cited by
-
Risk of epithelial ovarian cancer among women with benign ovarian tumors: a follow-up study
Cancer Causes & Control (2020)
-
Diagnosis and management of a giant ovarian cyst in the gravid-puerperium period: a case report
BMC Pregnancy and Childbirth (2019)
-
The disparate origins of ovarian cancers: pathogenesis and prevention strategies
Nature Reviews Cancer (2017)
-
Inhibition of nuclear factor-kappa B enhances the tumor growth of ovarian cancer cell line derived from a low-grade papillary serous carcinoma in p53-independent pathway
BMC Cancer (2016)
-
KRAS, BRAF genotyping reveals genetic heterogeneity of ovarian borderline tumors and associated implants
BMC Cancer (2013)
