
Abstract
Study Design:
A basic study using a spinal cord injury (SCI) model in rats.
Objectives:
The effect of mild hypothermic treatment on histological changes and motor function after a rat spinal cord compression injury was assessed.
Methods:
Mild spinal cord compression was performed at the eleventh thoracic vertebral level by a 20 g weight for 20 min. Rats in the mild hypothermic model were kept at a body temperature of 33 °C and rats in the normothermic group were kept at 37 °C for 1 h from beginning of compression. Motor function was evaluated by measuring the frequency of standing. Microglia were stained by isolectin B4 and observed in the compressed portion of the spinal cord. The amount of tumor necrosis factor-α (TNF-α) in the compressed spinal cord was measured by the ELISA method.
Results:
In the normothermic rats, microglia proliferated up to 72 h after the compression. Proliferation was substantially inhibited at 48 and 72 h after compression in the hypothermic rats. The motor function of the hypothermic rats improved at 48 and 72 h after the compression, whereas no improvement was seen in the normothermic rats. The amount of TNF-α in the compressed portion of the spinal cord was lower in hypothermic rats compared with normothermic rats throughout the experiment.
Conclusions:
These results suggest that hypothermic treatment is effective for the amelioration of delayed motor dysfunction via inhibition of microglial inflammatory responses.
Similar content being viewed by others


Introduction
It is difficult to recover neuronal function after serious central nervous system (CNS) damage. Severe disabilities remain even if a patient survives CNS damage such as brain infarction, cerebral hemorrhage or spinal cord injury (SCI). Such patients need rehabilitation and support in order to move, and may not be able to work for a long period of time. This situation is grievous not only for the patient himself, but also for the patient's family, both economically and psychologically. In addition, the social insurance system and the labor force have been negatively affected by the large number of patients with CNS damage. Therefore, the development of effective treatments for CNS damage is an urgent matter.
Recently, mild hypothermic treatment (about 33 °C) has been reported as an effective treatment for severe brain damage. The method entails keeping the patient under general anesthesia and body temperature between 33 and 34 °C for several days to two weeks after the onset of disease. This therapy, which has been applied to cerebral hemorrhage and other brain damages, reduces neuronal degeneration.1, 2 Hayashi et al.3 reported that hypothermic treatment enabled patients, who otherwise would have become brain dead, to reintegrate into the society.
Microglia are immune cells in the CNS. Under conditions such as ischemia4 and trauma,5 microglia proliferate and are activated. These pathologically activated microglia accelerate neuronal damage by releasing nitric oxide, superoxide and inflammatory cytokines such as TNF-α.6, 7, 8
Previously, we developed a model for mild spinal cord compression injuries using rats.9 In this model, microglia proliferated and were activated after spinal cord compression. The proliferated microglia produced TNF-α and iNOS. Furthermore, the time course of the loss of hind-limb function was quite similar to that of the microglia proliferation in the spinal cord after the compression injury.
In the present study, to clarify the mechanism of hypothermic treatment, we used our mild SCI model and tested two groups, one which received hypothermic treatment and the other which did not. We then evaluated the relationship between delayed motor function loss and the microglial pathological reaction including TNF-α release in the injured spinal cord.
Materials and methods
Animals
In total, 47 female Wister rats (3–4 month old, 250–350 g weight; Nippon Clea, Tokyo, Japan) were used for this research. Twenty rats were used for the hypothermic group (5 for behavior and 15 for histological analysis), 19 for the normothermic group (5 for behavior and 14 for histological analysis) and 8 for the sham group (5 for behavior and 3 for histological analysis).
Mild spinal cord compression model in rats
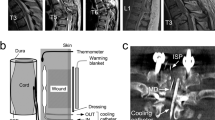
Previously, we developed a mild SCI model.9 Under general anesthesia using halothane, the rat's tenth and eleventh vertebral lamina were removed carefully without any damage to the spinal cord. Direct compression on the eleventh vertebral level was performed using a 20 g weight. The portion of the weight which contacted the dura was covered with soft, rounded silicone in order to prevent violent injury that would cause axonal tears or hemorrhage in the spinal cord (Figure 1). The weight was gently placed on the thoracic spinal cord extradurally for 20 min. Using this model, we did not observe any serious histological damage, such as hemorrhage or cavity formation in the spinal cord tissue. In the group of sham-operated rats, only laminectomies without compression were performed.

Method of the spinal cord injury model. The lamina of the eleventh and twelfth thoracic vertebrae was removed from rats anesthetized with halothane. The spinal cord was exposed enough to apply a weight. A 20 g weight was then gently placed on the exposed spinal cord. The point of the compression device was made of soft rounded silicone in order to prevent violent injury, which could cause axonal tears or hemorrhages in the spinal cord.
Control of rat body temperature
Rat body temperature was controlled using a water-cooling mat, and an animal body temperature controller with infrared ray irradiation (IFR-100, Unique Medical Co., Tokyo, Japan) (Figure 2). Water (31 and 35 °C) was continuously circulated in the water-cooling mat for the hypothermic and normothermic rats, respectively. The rats' rectal temperature was continuously monitored. When rectal temperature fell below 33 °C (hypothermic rats) or 37 °C (normothermic and sham rats), the animal body temperature controller detected this and the infrared ray irradiation was turned on automatically to increase the rats' body temperature. Using this system, the temperature of the animals was controlled for 1 h from the beginning of the compression period at 33 and 37 °C for the mild hypothermic and normothermic/sham rats, respectively. After the treatment, rats were returned to their in cages and body temperature was allowed to recover naturally.

Protocol for rat body temperature control. (a) Protocol for rat body temperature control in the hypothermic group. (b) Photograph of the rat body temperature control system. Temperature control was performed using a water-cooling mat and rat body temperature controller (IFR-100, Unique Medical Co.). (c) Schema of rat body temperature control system.
Evaluation of the rat behavior
Rat hind-limb function was evaluated by monitoring standing frequency using Scanet MV-10 (MATYS Co., Tokyo, Japan). Rats often assume a posture in which they lift their heads and forelimbs and support their weight only on the hind limbs. In the present paper, we call this vertical movement ‘standing’ (Figure 3). Rats were placed in a 480 mm × 480 mm box that had infrared ray detectors set 13 cm above the floor. The height of 13 cm was enough to detect only the standing posture; it did not detect the rats' movement when they raised their heads without lifting their forearms. The detector counted the number of times that the rats assumed the standing posture automatically. Rats usually assume this standing posture about 40–50 times per hour. A decrease in the standing frequency reflects the severity of motor dysfunction of the hind limbs induced by thoracic spinal cord damage.9 The standing frequency was measured for 1 h. Measurements were taken 24, 48 and 72 h after the spinal cord compression in a dark, silent room to prevent the influence of noise and light. Results were expressed as percentages of the average value from the sham rats (mean±s.e.m.). No significant difference was observed in standing frequency between normal (nonoperated) and sham-operated rats (data not shown).

Method of evaluating rat behavior. (a) Animal behavior was analyzed using Scanet MV-10 (MATYS Co.). (b) Hind-limb function was evaluated by measuring the frequency of vertical movement (standing) using infrared ray detectors, which were set 13 cm above the floor.
Histological examination
Rats were placed under deep anesthesia and sacrificed by decapitation. The compressed part of the spinal cord was dissected, and axial freezing microtome sections of 5 μm thickness were prepared on glass slides. The sections were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 5 min. Then, after washing twice with PBS, endogenous peroxidase was blocked by treatment with 0.3% H2O2/methanol for 15 min.
For microglial observation, lectin staining was performed. After pretreatment with 0.1% Triton-X (Wako Chemicals Ltd, Osaka, Japan) in PBS for 15 min, slices were exposed to peroxidase labeled-lectin from Bandeiraea simplicifolia (Isolectin B4, 5 μg ml−1 in PBS with 1% Triton X, SIGMA, St Louis, MO, USA) overnight at 4 °C. After washing with PBS for 20 min, sections were treated with 3,3′-diaminobenzidine tetrahydrochloride (DAB; Wako Chemicals Ltd) substrate (0.02% H2O2 plus 0.1% DAB in 0.1 M Tris-buffer) for 7 min and washed immediately with water for 20 min. Sections were dehydrated using graded alcohols and xylene, and were then mounted in HSR solution (Yoshitomi Co., Osaka, Japan). To quantify microglia proliferation, fine photographs were taken and the number of isolectin-positive cells in whole gray matter were counted by three individuals blind to information about the pictures. The observed numbers of isolectin-positive cells were averaged and data were expressed as the number of cells per square millimeter of gray matter. The area of the gray matter was calculated from the picture by using NIH Image software.
For TNF-α staining, endogenous peroxidase-blocked slices were washed with PBS for 5 min. The slices were exposed to anti-TNF-α antibody (1:1000; SIGMA) containing PBS with 1% horse serum at 4 °C overnight, and then washed with PBS for 20 min. Biotin-conjugated anti-mouse IgG (1:1000; SIGMA) in PBS with 1% horse serum was used as the secondary antibody. The slices were exposed to the secondary antibody for 30 min at room temperature. After washing the slices with PBS for 20 min, the abidin-biotin-complex technique (Vector, Burlingame, CA, USA) was performed. After washing with PBS for 20 min, sections were then treated with DAB substrate (0.02% H2O2 plus 0.1% DAB in 0.1 M Tris-buffer) for 7 min and washed immediately with water for 20 min. Sections were dehydrated and mounted in HSR solution.
Measurement of TNF-α content
At 24, 48 and 72 h after compression, the compressed portion of the rat spinal cord (1 cm in length) was taken and homogenized in 1 ml of ice cold PBS with proteinase inhibitor cocktail (1:100; SIGMA). About 20 μl of the sample was used for protein measurement. The remaining sample was centrifuged at 1000 × g for 15 min, and the supernatant was collected for TNF-α measurement. The amount of TNF-α was measured using an ELISA kit (BIOSOURSE, CA, USA) and the value was normalized by the protein content.
Data analysis
Statistic analyses of frequency of standing, number of lectin-positive cell numbers and TNF-α content were performed using JMP software (version 6.0.3J, 2005, SAS Institute, NC, USA). Mann–Whitney's U-test was used to compare between hypothermic and normothermic groups. P level of <0.05 was accepted as the significance level.
Statement of ethics
We certify that all applicable institutional and governmental regulations concerning the ethical use of animals were followed during the course of this research.
Results
Animal behavior
Standing frequency (hind-limb motor function) of the normothermic rats decreased throughout the experimental period after compression (39.1±13.4% (mean±s.e.m.), 36.2±16.8%, 28.7±15.6%, at 24, 48, 72 h after the compression, respectively). In hypothermic rats, no remarkable improvement was seen at 24 h after the compression (44.7±12.6% of sham rats). However, the standing frequency of the hypothermic rats at 48 h after compression significantly increased (86.6±23.8% of sham rats). Completely recovery (104.0±19.3%) of hind-limb function was observed at 72 h after compression. The frequency of standing in hypothermic rats at 48 and 72 h after compression was significantly higher than that in normothermic rats (P=0.0495 and 0.0495 for 48 and 72 h, respectively; Figure 4).

Effect of hypothermic treatment on rat behavior. A spinal cord compression procedure was performed in two groups of rats. Afterwards, the hypothermic and normothermic rats' body temperatures were maintained at 33 and 37 °C, respectively. Then, rat behavior was analyzed using Scanet MV-10 at 24, 48 and 72 h after the compression. Data are expressed as the mean±s.e.m. (n=6–7). Motor dysfunction in hypothermic rats was the same as that in normothermic rats at 24 h after compression. Motor function in hypothermic rats significantly increased to a normal level at 48 and 72 h after compression, however, no remarkable recovery was seen in normothermic rats.
Histological results
In the spinal cord tissue from a healthy rat, only a few isolectin-positive microglia were observed in a 5 μm-thick slice of spinal cord. In normothermic rats, microglia appeared 24 h after the compression in an area of 4.6±2.3 mm−2 (mean±s.e.m.; n=5) surrounding the compressed portion of the spinal cord. This area increased to 41.4±13 mm−2 (n=5) and 35.9±15.2 mm−2 at 48 and 72 h, respectively. In hypothermic rats, the number of microglia at 24 h after compression was similar to that observed in normothermic rats (4.5±5.0 mm−2). However, a significantly lower number of microglia were seen at 48 h (10.9±3.2 mm−2: P=0.0272) and 72 h (8.0±2.3 mm−2: P=0.0339) after compression compared with normothermic rats (Figure 5).

Lectin staining of injured spinal cord gray matter. Spinal sections were obtained at (a) 24, (b) 48 and (c) 72 h after spinal cord compression from normothermic (37 °C) and hypothermic (33 °C) rats. Then sections were stained with isolectin B4, which binds to microglia. (d) The coronal slices were stained with isolectin and the positive cells were counted. Data are expressed as the mean±s.e.m. (n=6–8). Microglial proliferation was significantly (P<0.05) inhibited at 48 and 72 h after compression in the hypothermic rats compared with normothermic rats.
Numerous TNF-α-positive cells were observed in normothermic rats and they were strongly stained at 48 h after the compression. On the other hand, in hypothermic rats, only a few poorly stained TNF-α-positive cells were observed (Figure 6).

TNF-α staining of normothermic and hypothermic rat spinal cord sections. A large number of TNF-α-positive cells were observed in the normothermic rat spinal cord slices (a). However, less TNF-α-positive cells were observed in the hypothermic rat slices and their intensity of staining was less than that of the normothermic rat slices (b).
TNF-α content
In the sham rats, the TNF-α concentration was 157.66±58.01, 199.13±149.38 and 257.66±122.85 (pg per mg protein, mean±s.d.) at 24, 48 and 72 h after the compression, respectively. In the normothermic rats, the TNF-α concentration was 484.70±40.25, 390.07±69.60 and 540.99±137.42 (pg per mg protein) at 24, 48 and 72 h after compression, respectively. TNF-α concentration in normothermic rats was always higher than those in the sham rats. In hypothermic rats, the TNF-α concentration was 306.01±114.04, 313.67±25.14 and 398.65±137.42 (pg per mg protein) at 24, 48 and 72 h after compression, respectively. No significant differences were detected between normothermic rats and hypothermic rats (P=0.2012, 0.9353 and 0.4649 for 24, 48 and 72 h after the compression, respectively), but the TNF-α concentration in hypothermic rats was observably less than that in normothermic rats through the experimental period (Figure 7).

TNF-α concentration in the compressed spinal cord at 24, 48 and 72 h after compression. The TNF-α concentration in hypothermic rats was less than that in normothermic rats at all the time points.
Discussion
In recent clinical studies, the effectiveness of mild hypothermic treatment, in which body temperature is reduced to 33 °C, has been reported for severe CNS damage such as brain trauma and ischemia.1, 2, 3, 10 Basic research to elucidate the mechanism of hypothermic treatment has also been performed. Hypothermia has been shown to inhibit the elevation of excitatory amino acid concentration in the brain after ischemia,11 neuronal apoptosis after spinal cord injuries in rabbits12 and post-traumatic inflammatory cascades such as IL-1β, iNOS and superoxide in the brain.13, 14 Moreover, hypothermia decreases intracranial pressure, increases microcirculation15 and preserves the blood–brain barrier against ischemic brain damage.16 These reports suggest that hypothermic treatment may act on delayed mechanisms of CNS damage, and encourage the possibility of clinical use of hypothermic treatment after traumatic or ischemic CNS injury. Inhibition of delayed neuronal damage after primary neuronal damage in the CNS has been believed to be an important therapeutic strategy to minimize neuronal damage and may improve the clinical outcome in patients with CNS damage.
Microglia, immune cells in the CNS, have been reported to proliferate after CNS damage.4 These proliferated microglia possibly accelerate neuronal damage by releasing toxic substances.6, 7, 8 We previously reported that microglial proliferation and activation were observed in the damaged spinal cord, and that the time course of the progress of hind-limb function loss was quite similar to that of the microglia proliferation in spinal cord after a compression injury. The proliferated microglia were positively stained by anti-TNF-α and anti-iNOS antibodies.9 We concluded that the delayed motor dysfunction is related to microglial proliferation via its inflammatory responses. Since inflammatory responses continue several days after CNS damage, inhibitory therapy for such delayed mechanisms, including microglial proliferation, should be effective even after the acute phase in SCI. There are several reports which state that hypothermic treatment is effective even when it starts after the ischemic brain damage17 and SCI.18
In the present study, hypothermic treatment decreased TNF-α production from activated microglia. Westmoreland et al.19 have reported that the addition of TNF-α induced cell death in cultured human neuronal cell lines. Pentoxifylline, which inhibits the effects of TNF-α, had a significant protective effect in this assay. Zhang and Fedoroff20 reported that neurons in mixed neuron-microglia cultures survived longer than those in pure neuron cultures, however, when they treated the mixed culture with lipopolysaccharide, an activator of immune cells, the microglia impeded neuronal survival. Viviani et al.21 reported that activation of TNF-α release from glial cells induced neuronal death, which was inhibited by the addition of TNF-α ntibody in glia–neuron cocultures. These papers indicate that TNF-α produced by activated microglia is possibly toxic to neurons and that inhibition of pathological microglial function is an effective therapeutic strategy against secondary neuronal damage after traumatic SCI.
In the present paper, delayed motor dysfunction up to 72 h after the SCI was ameliorated by 33 °C hypothermic treatment. Parallel with this improvement, microglial proliferation and TNF-α release were inhibited. These results suggest that moderate hypothermic treatment after a SCI reduces delayed motor dysfunction. Inhibition of pathological microglial proliferation and activation are important factors in the mechanism of hypothermic treatment for CNS damage.
References
Kataoka K, Yanase H . Mild hypothermia—a revived countermeasure against ischemic neuronal damages. Neurosci Res 1998; 32: 103–117.
Bernard S . Induced hypothermia in intensive care medicine. Anaesth Intensive Care 1996; 24: 382–388.
Hayashi N, Hirayama T, Udagawa A, Daimon W, Ohata M . Systemic management of cerebral edema based on a new concept in severe head injury patients. Acta Neurochir Suppl 1994; 60: 541–543.
Morioka T, Kalehua AN, Streit WJ . The microglial reaction in the rat dorsal hippocampus following transient forebrain ischemia. J Cereb Blood Flow Metab 1991; 11: 966–973.
Watanabe T, Yamamoto T, Abe Y, Saito N, Kumagai T, Kayama H . Differential activation of microglia after experimental spinal cord injury. J Neurotrauma 1999; 16: 255–265.
Meda L, Cassatella MA, Szendrei GI, Otvos Jr L, Baron P, Villalba M et al. Activation of microglial cells by β-amyloid protein and interferon-γ. Nature 1995; 374: 647–650.
Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK . Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol 1992; 149: 2736–2741.
Tanaka M, Sotomatsu A, Yoshida T . Detection of superoxide production by activated microglia using a sensitive and specific chemiluminescence assay and microglia-mediated PC 12 h cell death. J Neurochem 1994; 63: 266–270.
Morino T, Ogata T, Horiuchi H, Takeba J, Okumura H, Miyazaki T et al. Delayed neuronal damage related to microglia proliferation after mild spinal cord compression injury. Neurosci Res 2003; 46: 309–318.
Adelson PD, Ragheb J, Kanev P, Brockmeyer D, Beers SR, Brown SD et al. Phase II clinical trial of moderate hypothermia after severe traumatic brain injury in children. Neurosurgery 2005; 56: 740–754.
Busto R, Dietrich WD, Globus MY, Valdes I, Scheinberg P, Ginsberg MD . Small differences in intraischemic brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab 1987; 7: 729–738.
Lei MW, Ye Y, Liang JZ, Nai HJ, Zhi YXU . Moderate hypothermia prevents neural cell apoptosis following spinal cord ischemia in rabbits. Cell Res 2005; 15: 387–393.
Dietrich WD, Chatzipanteli K, Vitarbo E, Wada K, Kinoshita K . The role of inflammatory processes in the pathophysiology and treatment of brain and spinal cord trauma. Acta Neurochir Suppl 2004; 89: 69–74.
Luo J, Li N, Robinson JP, Shi R . The increase of reactive oxygen species and their inhibition in an isolated guinea pig spinal cord compression model. Spinal Cord 2002; 40: 656–665.
Burger R, Vince GH, Meixensberger J, Roosen K . Hypothermia influences time course of intracranial pressure, brain temperature, EEG and microcirculation during ischemia-reperfusion. Neurol Res 1998; 20: s52–s60.
Diretrich WD, Busto R, Halley M, Valdes I . The importance of brain temperature in alterations of the blood-brain barrier following cerebral ischemia. J Neuropathol Exp Neurol 1990; 49: 486–497.
Busto R, Dietrich WD, Globus MY, Ginsberg MD . Postischemic moderate hypothermia inhibits CA1 hippocampal ischemic neuronal injury. Neurosci Lett 1989; 101: 299–304.
Tsutsumi K, Ueda T, Shimizu H, Hashizume K, Yozu R . Effect of delayed induction of postischemic hypothermia on spinal cord damage induced by transient ischemic insult in rabbits. Jpn J Thorac Cardiovasc Surg 2004; 52: 411–418.
Westmoreland SV, Kolson D, Gonzalez-Scarano F . Toxicity of TNF alpha and platelet activating factor for human NT2N neurons: a tissue culture model for human immunodeficiency virus dementia. J Neurovirol 1996; 2: 118–126.
Zhang SC, Fedoroff S . Neuron-microglia interactions in vitro. Acta Neuropathol 1996; 91: 385–395.
Viviani B, Corsini E, Galli CL, Marinovich M . Glia increase degeneration of hippocampal neurons through release of tumor necrosisfactor-alpha. Toxicol Appl Pharmacol 1998; 150: 271–276.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Morino, T., Ogata, T., Takeba, J. et al. Microglia inhibition is a target of mild hypothermic treatment after the spinal cord injury. Spinal Cord 46, 425–431 (2008). https://doi.org/10.1038/sj.sc.3102163
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.sc.3102163
