
Abstract
Prepulse inhibition (PPI) of the acoustic startle response is an operational measure of sensorimotor gating that can be assessed in animals and in humans. Serotonin releasers such as MDMA disrupt PPI and reduce startle habituation in rodents. These effects are prevented by pretreatment with selective serotonin uptake inhibitors, indicating that the effect of MDMA on startle plasticity is largely due to carrier-mediated release of serotonin from presynaptic terminals. In contrast, MDMA has been shown to increase PPI in humans. It is unclear, however, whether the MDMA-induced increase in PPI in humans is also dependent on carrier-mediated serotonin release and which postsynaptic receptors are involved. We investigated the effects of three different pretreatments on the MDMA-induced effects on PPI and habituation in humans. Pretreatments were: (1) the highly selective serotonin uptake inhibitor citalopram (40 mg IV) in 16 subjects, (2) the D2 antagonist haloperidol (1.4 mg IV) in 14 subjects, and (3) the 5-HT2A/C antagonist ketanserin (50 mg PO) in 14 subjects. Each of the three studies used a double-blind placebo-controlled design. All healthy volunteers were examined four times at 2–4-week intervals after placebo, pretreatment, MDMA (1.5 mg/kg PO), and pretreatment plus MDMA. MDMA increased PPI. Habituation was not altered by MDMA, although MDMA-induced individual differences on habituation and psychological symptoms were inversely correlated. Citalopram attenuated the MDMA-induced increase in PPI and most of the psychological effects of MDMA. Neither haloperidol nor ketanserin had any effect on PPI increases produced by MDMA, although each partially attenuated some MDMA-induced psychological effects. Results are consistent with the view that MDMA increases PPI of the acoustic startle reflex in humans via release of presynaptic serotonin.
Similar content being viewed by others

Main
The startle reflex is a contraction of the skeletal and facial musculature in response to a sudden intense stimulus, such as a loud noise. In humans, the eyeblink component of the startle reflex is measured using electromyography of the orbicularis oculi muscle. In rodents, a stabilimeter is used to register a whole-body flinch response. The startle reflex exhibits several forms of behavioral plasticity, such as prepulse inhibition (PPI) and habituation, which are consistent phenomena across species. PPI refers to the normal suppression of the startle response when the startling stimulus is preceded by a weak prestimulus. PPI is regarded as an operational measure of sensorimotor gating reflecting the ability to filter cognitive or sensory information. Habituation refers to the decrement of the startle response observed with repeated presentation of the stimulus and is considered to be the simplest form of learning and a prerequisite of selective attention. Both PPI and habituation deficits have been found in patients with schizophrenia (Bolino et al. 1994; Braff et al. 1992; Geyer and Braff 1982) and schizotypal personality disorder (Cadenhead et al. 1993). Deficits in PPI are also seen in obsessive–compulsive disorder (Swerdlow et al. 1993) and Huntington's disease (Swerdlow et al. 1995).
In animals, MDMA (3,4-methylenedioxy-N-methylamphetamine, “Ecstasy”) releases serotonin (5-HT) and, to a lesser extent, dopamine. The pharmacological effects of MDMA-like drugs on PPI are well studied in rodents [for a review see (Geyer and Callaway 1994)]. Entactogens including MDMA, MDEA (N-ethyl-3,4-methylenedioxyamphetamine), AET (alpha-ethyltryptamine), as well as the serotonin releaser fenfluramine, impair both PPI and habituation of the startle reflex in rodents (Dulawa and Geyer 1996; Kehne et al. 1996; Mansbach et al. 1989; Martinez and Geyer 1997; Vollenweider et al. 1999a). The effects of MDMA-like drugs on PPI and startle habituation are reduced by pretreatment with selective serotonin uptake inhibitors, which prevent carrier-mediated release of presynaptic serotonin by MDMA (Kehne et al. 1992; Martinez and Geyer 1997). These findings support the hypothesis that the effects of MDMA on PPI and habituation are mediated via release of endogenous serotonin. In addition, the 5-HT2A antagonist MDL 100,907 (now M100907) also reduces the effect of MDMA on PPI (Padich et al. 1996), indicating that the released serotonin affects PPI by stimulating postsynaptic 5-HT2A receptors. Consistent with this conclusion, direct 5-HT2A receptor activation has been found to disrupt PPI in rats and this effect is reversed by 5HT2A antagonists (Sipes and Geyer 1994, 1997). In addition, both 5-HT1A and 5-HT1B agonists reduce PPI in rats and these effects are antagonized by the corresponding receptor antagonists (Rigdon and Weatherspoon 1992; Sipes and Geyer 1994,1995). Thus, serotonin released by MDMA might also disrupt PPI in rats by acting at 5-HT1A and/or 5-HT1B receptor sites. MDMA also releases dopamine and the dopamine releaser d-amphetamine as well as dopamine agonists disrupt PPI (Mansbach et al. 1988; Sills 1999; Swerdlow et al. 1990). However, the D2 antagonist haloperidol did not prevent the MDMA-induced disruption of PPI (Padich et al. 1996). In sum, these data from animal studies indicate that MDMA affects PPI by activating the serotonin system.
Little is known about the pharmacology of PPI in humans. Surprisingly, a recent study from our laboratory found that MDMA (1.7 mg/kg PO) increased PPI in healthy volunteers, thus having an opposite effect on sensorimotor gating in humans versus animals (Vollenweider et al. 1999a). Whether procedural or species-specific differences, including a different mechanism of action of MDMA, account for these findings is unclear.
Therefore, the aims of the present study were several. First, the previous findings of an MDMA-induced increase in PPI should be confirmed. Second, three pretreatments—the highly selective serotonin uptake inhibitor citalopram, the D2 antagonist haloperidol, and the 5-HT2A/C antagonist ketanserin—were used in order to prevent MDMA effects on PPI to the extent that they depend on: (1) MDMA-induced carrier-mediated release of presynaptic serotonin, (2) D2 stimulation, or (3) 5-HT2A/C stimulation. It was hypothesized that citalopram would attenuate the effect of MDMA on PPI, based on the fact that selective serotonin uptake inhibitors reduce MDMA-induced serotonin release (Gudelsky and Nash 1996; Hekmatpanah and Peroutka 1990; Koch and Galloway 1997) and behavioral effects in animals (Callaway et al. 1990). Third, correlations should be assessed between differences in MDMA-induced psychological changes and changes in %habituation or %PPI.
Materials and methods
Participants
The study was approved by the Ethics Committee of the University Hospital of Psychiatry, Zurich, and the use of MDMA was authorized by the Swiss Federal Health Office, Department of Pharmacology and Narcotics, Bern. Fifty healthy volunteers were recruited from University Hospital staff and at the Medical School of the University of Zurich. All volunteers gave their written consent after being informed by a written and oral description of the aim of the study, the procedures involved, and the effects and possible risks of MDMA administration. Of the 44 volunteers included in the study, 42 were either university students or physicians. Subjects were healthy according to medical history, clinical examination, electrocardiography and blood analysis, and were screened by psychiatric interview to exclude those with personal or family (first-degree relatives) histories of major psychiatric disorder. Subjects with regular alcohol or substance abuse were excluded. Some participants had minimal prior drug experiences (once or twice, all more than 6 months prior to the study); all other subjects were drug-naıuml;ve. Previous drug experiences were as follows. Of the 16 subjects included in the citalopram–MDMA study, 8 had tried cannabis, 2 MDMA, and 3 an hallucinogen; 2 were light smokers. Of the 14 subjects included in the haloperidol–MDMA study, 7 had tried cannabis, 1 MDMA, 3 an hallucinogen; 2 were light smokers. Of the 14 subjects included in the ketanserin–MDMA study, 7 had tried cannabis, 2 MDMA, 4 an hallucinogen, and there were no smokers. None of the subjects had used amphetamine or cocaine. All subjects underwent a pretest startle session to assure startle responsiveness prior to admission to the study. Of the 50 candidates, 6 had to be excluded due to psychiatric disorder, recreational drug use, or insufficient startle response. All 44 volunteers included in this study were measured four times and all received MDMA, placebo, one of three different pretreatments prior to MDMA, or the pretreatment alone. Of the 44 subjects, 16 received the selective serotonin uptake inhibitor citalopram as pretreatment (12 male, 4 female; mean age 27 years, range 21–39); 14 received the D2 dopamine antagonist haloperidol as pretreatment (9 male, 5 female; mean age 26 years, range 21–38); 14 subjects received the 5-HT2A/C antagonist ketanserin as pretreatment (13 male, 1 female; mean age 26 years, range 21–41). For the pooled analysis of MDMA effects compared to placebo, psychometric data were available from 44 subjects, and startle data from 38 subjects (31 male and 7 female). Analysis of startle data of the citalopram–MDMA study included 14 subjects (10 male, 4 female); analysis of the haloperidol–MDMA study included 12 subjects (9 male, 3 female); analysis of the ketanserin–MDMA study also 12 subjects (12 males); due to 2 drop-outs in each study due to insufficient startle responses or artifacts.
Drugs and Dosing
Racemic MDMA was obtained through the Swiss Federal Health Office, Department of Pharmacology and Narcotics, Berne, and prepared as capsules of 10 and 50 mg. Subjects received MDMA at a moderate dose of 1.5 mg/kg PO. The mean absolute dose was 103 mg, with an absolute dose range from 70–120 mg. This dose of MDMA is known to have robust psychological effects and to significantly increase PPI in healthy humans (Vollenweider et al. 1998a). The dose was also carefully evaluated to minimize possible risks and is unlikely to produce lasting harmful effects (Lieberman and Aghajanian 1999; Vollenweider et al. 1999b). It was decided not to use multiple doses of MDMA for this within-subject study, because such a design would require multiple administrations of MDMA and order effects might occur. Thus, one dose was used twice in this four-session cross-over study in order to limit the exposure to MDMA within the same subject.
Citalopram is the most selective SSRI currently available (Hyttel et al. 1995). Citalopram hydrochloride ampules, 40 mg, were kindly provided by Lundbeck, Switzerland. Citalopram (40 mg) was dissolved in 500 ml sterile saline solution and given slowly by IV perfusion over 90 min (330 ml/h) in compliance with the Swiss drug registration council's recommendations. Because PO citalopram has a bioavailability of almost 100%, the IV dose used here is equivalent to a PO dose of 40 mg, which is a usual clinical starting dose (20–40 mg) (Baumann and Larsen 1995; Milne and Goa 1991). IV administration of citalopram has been shown to produce similar side effects as PO administration in depressed patients (Baumann et al. 1998). Tolerability was also favorable in seven of eight healthy volunteers (Seifritz et al. 1996) and in pilot studies conducted in our laboratory using 40–80-mg citalopram infusions.
Haloperidol (1.4 mg) was given IV 10 min prior to MDMA. This dose is equivalent to a PO dose of about 2.8 mg (Froemming et al. 1989), was expected to occupy about 60% of dopamine D2 receptors (Nordström et al. 1992), and has been shown previously to antagonize some psychological effects of the 5-HT2/1 agonist psilocybin using a comparable design (Vollenweider et al. 1998b).
Ketanserin was kindly provided by Janssen-Cilag, Switzerland, and was prepared as gelatine capsules of 50 mg and given PO 75 min prior to MDMA. The same time conditions and a similar dose have been shown previously to robustly antagonize most psychological effects of psilocybin (Vollenweider et al. 1998b).
Experimental Protocol
A double-blind placebo-controlled within-subject design was used for each study with four experimental conditions: placebo–placebo; pretreatment–placebo; placebo–MDMA; pretreatment–MDMA (with order being counterbalanced). Thus, each of the 16 participants in the citalopram study received placebo, citalopram, MDMA, and citalopram plus MDMA on four different days. In identical designs, each of the 14 participants in the haloperidol–MDMA study received placebo, haloperidol, MDMA and haloperidol plus MDMA, and each of the 14 participants in the ketanserin–MDMA study received placebo, ketanserin, MDMA and ketanserin plus MDMA. For the male subjects, the sessions were separated by at least 2 weeks to minimize carry-over effects. The female subjects were tested at monthly intervals, at the same day of their menstrual phase to minimize gender differences and influences of the menstrual cycle on PPI (Swerdlow et al. 1996). Sessions were conducted in a calm and comfortable laboratory environment. Participants were told to abstain from alcohol the day prior to each session and not to drink caffeinated beverages or to eat 2 h prior to each session. The four light smokers were told to maintain their usual smoking habits, but did not smoke during sessions. At the beginning of each session in the citalopram–MDMA study, citalopram (40 mg in 500 ml saline solution) or placebo (500 ml saline solution alone) was infused over 90 min. MDMA was given (1.5 mg/kg PO) immediately after removal of the IV line. Volunteers in the haloperidol-MDMA study received an IV injection of either haloperidol (1.4 mg) in saline or saline alone. After 10 min, MDMA (1.5 mg/kg) or placebo was given PO. In the ketanserin study, the time interval between intake of ketanserin (50 mg PO) and MDMA (1.5 mg/kg PO) was 75 min. Sensorimotor gating of the acoustic startle reflex was measured 90 min after MDMA/placebo intake (i.e., about 30–45 min after the expected onset of subjective effects of MDMA). Mood ratings were performed 120 min after MDMA/placebo intake, about 60 to 75 min after the expected onset of subjective effects. Blood pressure, heart rate, and body temperature were monitored throughout the session. Side effects of all drug conditions were assessed by the List of Complaints (von Zerssen 1976). Acute effects were assessed during the session, short-term sequelae the next day (24 h) and again 3 days after the test session (72 h). After the acute effects of MDMA had subsided, subjects remained in the hospital for another 2 h and were monitored clinically. After discharge subjects could contact the researchers at any time in case of adverse drug effects or psychological problems. At the end of each study subjects attended a debriefing and follow-up interview. All these results are presented in detail elsewhere (Liechti et al. 2000b; Liechti and Vollenweider 2000a,b).
Psychological Ratings
The Adjective Mood rating scale (AM) (Janke and Debus 1978) and the Altered State of Consciousness (OAV) rating scale (Dittrich et al. 1985; Dittrich 1998) were used to assess psychological peak drug effects of the four treatment regimens. The AM and OAV scales had previously been shown to be sensitive to psychological effects of MDMA in humans (Vollenweider et al. 1998a).
The AM questionnaire consists of 14 scales measuring “efficiency-activation,” “self-confidence,” “heightened mood,” “apprehension-anxiety,” “depressiveness,” “thoughtfulness-contemplativeness,” “extroversion,” “introversion,” “inactivation,” “dazed state,” “tiredness,” “sensitivity,” “aggression-anger,” and “emotional excitation.”
The OAV rating scale is a visual-analogue scale that measures alterations in waking consciousness, including changes in mood, perception, experience of oneself and of the environment, as well as thought disorder. The OAV questionnaire consists of three scales comprising several item clusters. (1) OB (“oceanic boundlessness”), measures derealization and depersonalization associated with positive mood ranging from heightened feelings to exaltation and alterations in the sense of time. The corresponding item clusters are “derealization,” “depersonalization,” “alterations in the sense of time,” “positive basic mood” and “mania-like experience.” (2) AED (“anxious ego dissolution”), measures ego-disintegration and loss of autonomy and self-control associated with arousal and anxiety. The item clusters are “thought disorder,” “fear of loss of thought control,” “fear of loss of body control,” “anxious derealization,” and “delusion.” (3) VR (“visionary restructuralization”), includes the item clusters “elementary hallucinations,” “visual (pseudo-) hallucinations,” “synesthesia,” “changed meaning of percepts,” “facilitated recollection,” and “facilitated imagination.”
Apparatus and Startle Test Session
Subjects were seated comfortably in an arm-chair and were instructed to keep their eyes open. The eyeblink component of the acoustic startle response was measured using an electromyographic startle system (EMG-SR-LAB, San Diego Instruments, Inc., San Diego, CA) and registration parameters as described in detail elsewhere (Braff et al. 1992). Two silver/silver chloride electrodes were placed below the right eye over the orbicularis oculi muscle and a ground electrode was placed behind the right ear. All electrode resistances were less than 5 kΩ. Acoustic startle stimuli were presented through headphones (TDH-39-P, Maico). Each session began with a 5-min acclimation period of 70-dB background white noise that continued throughout the session. The session consisted of 60 trials consisting of three conditions: (1) a 115-dB pulse alone (PA) of 40 ms duration; (2) the same pulse preceded by a 16-dB (above background) prepulse of 20 ms duration at 30, 60, 120, 240 or 2000 ms (PP30, PP60, PP120, PP240, PP2000); and (3) no-stimulus trials (NS). Sessions were analyzed in four blocks (time intervals). The first and last blocks of a session consisted of six PA trials each, that were not used for the calculation of PPI. The middle two blocks of in sum 48 trials consisted of 12 PA, and 6 each of the PP30, PP60, PP120, PP240, PP2000 and NS trials presented in a pseudorandom order. NS trials were used to detect base-line shifts. The entire test session lasted about 18 min. All recordings were screened to exclude spontaneous eyeblink activity prior to data analysis, and about 5% of trials were excluded using previously described criteria (Braff et al. 1992).
Statistical Analysis
Startle magnitude was assessed for each block of a session using mean values of all PA trials. Percent habituation was calculated as the reduction in startle magnitude between the first and the last blocks of PA trials [%HAB = 100 × (magnitude first block – magnitude last block)/magnitude first block]. %PPI was calculated for each prepulse trial condition (PP30, PP60, PP120, PP240, PP2000) as the reduction in startle magnitude in the presence of a prepulse compared to the magnitude of the PA response [%PPI = 100 × (magnitude PA − magnitude prepulse plus pulse trial)/magnitude PA]. %PPI was calculated because PPI expressed as percentage is typically independent of the magnitude of the startle response as compared to PPI expressed as absolute difference score (Abel et al. 1998; Schwarzkopf et al. 1993).
Data were analyzed using STATISTICA 5.4 (StatSoft™) for Windows. Possible order effects (e.g., MDMA first vs placebo first, and MDMA first vs pretreatment-MDMA first) were excluded using two-way ANOVA with drug and order as factors for each dependent variable. The effect of MDMA on startle reactivity was analyzed using two-way ANOVA with MDMA (placebo vs MDMA) and startle magnitude (first, middle, and last block of the session) as repeated measures. The effects of MDMA on percent PPI were assessed using two-way ANOVA with MDMA (placebo vs MDMA) and PPI condition (PP30, PP60, PP120, PP240, PP2000) as repeated measures. To test whether citalopram, haloperidol, or ketanserin pretreatment blocked the MDMA-induced changes in PPI, we used three-way ANOVA with pretreatment (placebo vs citalopram, haloperidol, or ketanserin), MDMA (placebo vs MDMA), and PPI condition (PP60, PP120, PP240) as within-subject factors. Post hoc analyses were done using LSD tests based on significant main effects or interactions. Greenhouse-Geisser adjustments were applied to correct for the multiple levels of the repeated measures factors.
Psychometric scores were compared by ANOVA with MDMA (placebo vs MDMA) as the within-subject factor for the pooled data (n = 44). To assess the effects of pretreatments with citalopram, haloperidol, or ketanserin, two-way ANOVAs were used with pretreatment (citalopram, haloperidol, or ketanserin vs placebo) and treatment (placebo vs MDMA) as within-subject factors.
Spearman's rank-order correlations were used to assess the relationship between psychological peak changes (MDMA values – placebo values) and MDMA-induced changes in %habituation or %PPI (60-ms, 120-ms, and 240-ms intervals separately) (MDMA values − placebo values) in 43 subjects. The criterion for significance was set at p < .05.
Results
Psychological Effects of MDMA Compared to Placebo
Subjective effects of MDMA began 45–60 min after drug intake, peaked at about 90–120 min, and lasted for a mean duration of 3.5 h. Table 1 shows psychometric results for the MDMA and placebo conditions of all 44 subjects.
MDMA increased scores in all scales of the OAV questionnaire, OB [F(1,43) = 61.24; p < .001], AED [F(1,43) = 31.44; p < .001], and VR [F(1,43) = 18.85; p < .001]. MDMA-induced elevation in OB scores was due mainly to increases in “positive mood,” positively experienced “depersonalization,” and “mania-like experience” (all p < .001). The increase in AED scores was due mostly to “thought disorder,” and slight “fear of loss of body control” and “fear of loss of thought control” (all p < .001). Thought disturbances were moderate and included difficulty concentrating, accelerated thinking, thought blocking, and impaired decision making. Increases in VR scores were mainly attributable to “changes in the meaning of percepts,” “facilitated recollection,” and “facilitated imagination.” No hallucinations were reported, but subjects described an intensification of sensory perception. Typically, colors were described to be more vivid, touch was altered, and sound seemed closer or farther away.
In the AM mood rating scale, MDMA markedly increased “self-confidence” [F(1,43) = 24.21; p < .001], “heightened mood” [F(1,43) = 27.82; p < .001] and both “extroversion” [F(1,43) = 26.65; p < .001] and “introversion” [F(1,43) = 7.34; p < .01]. MDMA increased “sensitivity” [F(1,43) = 9.98; p < .01] and “emotional excitation” [F(1,43) = 33.45; p < .001] but also heightened “thoughtfulness-contemplativeness” [F(1,43) = 41.46; p < .001]. MDMA also increased scores for “inactivation” [F(1,43) = 4.70; p < .04] and “dazed state” [F(1,43) = 49.45; p < .001]. In contrast, MDMA nonsignificantly reduced “tiredness” compared to placebo [F(1,44) = 2.80; p < .1]. Although female subjects tended to have higher scores on some dependent measures, there were no significant gender differences in subjective responses to MDMA in this study.
Acute side effects of MDMA were “difficulty concentrating,” “dry mouth,” “impaired balance,” “dizziness,” “jaw clenching,” and “lack of appetite” in more than half of the subjects. The cardiovascular response to MDMA included elevated systolic and diastolic blood pressure (mean changes of plus 30 mm Hg and plus 15 mm Hg, respectively), and an increase in heart rate by about 12 beats per min (for details see Liechti et al. 2000b; Liechti and Vollenweider 2000a,b).
Startle Measures MDMA Compared to Placebo
Habituation was similar after placebo (mean ± SD) 29.38 ± 18.60 and after MDMA 29.32 ± 20.93. Similar habituation was also confirmed for MDMA and placebo by two-way ANOVA with MDMA and block and the absence of a drug × block interaction. MDMA nonsignificantly increased startle reactivity compared to placebo. ANOVA with gender as an additional factor revealed no significant differences between male and female subjects.
Figure 1 shows %PPI scores after placebo and MDMA administration. Prepulse effects were confirmed by ANOVA with drug and prepulse condition as factors. There was a significant main effect of prepulse condition [F(3.1,115) = 57.88; p < .001], confirming the dependence of PPI on the intervals. The main effect of MDMA was significant [F(1,37) = 10.69; p < .01], confirming that MDMA increased PPI compared to placebo. ANOVA including gender as an additional between-subject factor revealed that male subjects showed slightly higher PPI than female subjects [gender × PPI interaction: F(4,144) = 2.83; p < .03], and that the effect of MDMA on PPI was similar in both genders. Post hoc analyses revealed that the MDMA-induced increase in PPI was significant for the 60-ms (p < 0.01), 120-ms (p < .05), and 240-ms (p < .001) intervals, but not for the 30-ms interval. The PP2000 (2000-ms interval) produced prepulse facilitation both after placebo (−12.96 ± 23.50) and MDMA (−3.15 ± 16.38). Prepulse facilitation was decreased (p < .05) after MDMA compared to placebo. Because MDMA effects were generally similar for all prepulse intervals, as indicated by the lack of a significant drug × prepulse condition interaction, values from those PPI conditions that were significantly increased by MDMA were collapsed to a mean %PPI score (mean of PP60, PP120, and PP240).

Mean and SE values for percent PPI after placebo and MDMA. MDMA significantly increased PPI as indicated by *p < .05, **p < .01, and ***p < .001 (ANOVA and LSD post hoc tests)
Effects of Pretreatments with Citalopram, Haloperidol, or Ketanserin on MDMA-induced Psychological Changes
OAV scores are shown in Figure 2 . As previously shown in detail (Liechti et al. 2000a,b; Liechti and Vollenweider 2000b), all three pretreatments (citalopram, haloperidol, and ketanserin) significantly reduced several MDMA-induced effects and in addition showed different patterns in reducing effects of MDMA. Citalopram was the most effective pretreatment, reducing MDMA-induced changes in all three scales (OB, AED, and VR) of the OAV questionnaire [pretreatment × treatment interactions: OB F(1,15) = 22.47; p < .001; AED F(1,15) = 23.04; p < .001; VR F(1,15) = 12.80; p < .03]. In contrast, haloperidol reduced only MDMA-induced increases in OB scores [pretreatment × treatment interaction: F(1,13) = 5.89; p < .03], due to decreased “positive mood” and “mania-like experience,” but was ineffective in reducing AED or VR scores. Similarly, ketanserin reduced only MDMA-induced VR scores [pretreatment × treatment interaction: F(1,13) = 15.82; p < .01], indicating a reduction of MDMA-induced perceptual changes, but had no significant effect on OB or AED scores.

Mean and SE of t-transformed OAV scores of the Altered States of Consciousness Questionnaire (OAV). OB = oceanic boundlessness, AED = anxious ego-dissolution, VR = visionary restructuralization. P = placebo, C = citalopram, H = haloperidol, K = ketanserin, and M = MDMA. Significant MDMA-induced changes are indicated by *p < .05, **p < .01, and ***p < .001 compared to placebo (ANOVA, drug main effects). Significant reductions of MDMA-induced scores by pretreatment are indicated by #p < .05, and ###p < .001 (two-way ANOVA, pretreatment × treatment interaction)
All pretreatments also produced some adverse effects. Citalopram mainly produced nausea (6/16) compared to placebo (0/16). Drowsiness was the major complaint after haloperidol (11/14) compared to placebo (2/14) and after ketanserin (8/14) compared to placebo (7/14).
Startle Measures, Citalopram, and MDMA
Habituation refers to the decrease in startle magnitude between the first and the last blocks of a session, which is illustrated for all drug conditions in Figure 3 . Three-way ANOVA with MDMA, citalopram, and block as factors revealed a significant main effect of block [F(1.7,22) = 53.41; p < .001], confirming startle habituation. There were no differences in habituation between drug conditions. As seen in Figure 3, both MDMA and citalopram increased startle magnitude. Two-way ANOVA with drug (MDMA vs placebo; citalopram vs placebo) as factors confirmed that citalopram alone significantly increased startle magnitude compared to placebo [main effect of drug: F(1,13) = 8.0; p < .02]. The MDMA-induced increase in startle reactivity did not reach significance. However, there was a significant citalopram × MDMA interaction using three-way ANOVA with MDMA, citalopram, and block as factors [F(1,13) = 5.43; p < .04], indicating that the increase in startle reactivity induced by citalopram alone and the slight increase by MDMA did not sum up when the two drugs were taken together.

Mean and SE values of startle magnitudes of the first and the last blocks of the session. Habituation refers to the decrement in startle magnitude between the first and the last block. MDMA had no significant effect on startle magnitude. Citalopram increased startle magnitude in the first block, *p < .1, and in the last block (**p < .01). Haloperidol had no effect on startle magnitude. Ketanserin reduced startle magnitude, which was significant in the last block (**p < .01). There was no such reduction when ketanserin and MDMA were given together
Citalopram did not significantly change PPI compared to placebo as confirmed by ANOVA.
Two-way ANOVA with MDMA (MDMA vs placebo) as a within-subject factor revealed that MDMA significantly increased %PPI compared to placebo in this study [main effect of drug: F(1,13) = 5.27; p < .039]. This increase was most prominent and significant for the 60 ms [F(1,13) = 7.75; p < .015] and the 240 ms [F(1,13) = 9.96; p < .008] intervals, but did not reach significance for the 120-ms interval.
As shown in Figure 4 , the effect of MDMA on PPI was reduced by citalopram pretreatment which was confirmed by three-way ANOVA with citalopram, MDMA, and PPI condition as within-subjects factors. Only those prepulse conditions were included that were increased by MDMA (PP60, PP120 and PP240). There was a significant citalopram × MDMA interaction [F(1.13) = 6.3; p < .026], in the absence of significant main effects of drug. Neither citalopram nor MDMA interacted significantly with the prepulse condition. Pairwise two-way ANOVAs confirmed the significant citalopram × MDMA interaction for the 240-ms interval [F(1,13) = 11.7; p < .005], and indicated a trend for a significant interaction for the 60-ms interval [F(1,13) = 3.63; p < .08], but not for the 120-ms interval. There were no gender differences.

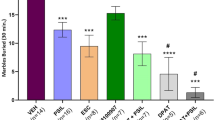
Mean and SE values of mean percent prepulse inhibition (60-, 120-, and 240-ms intervals collapsed) of the startle response after placebo (P), citalopram (C), MDMA (M), citalopram plus MDMA (CM), haloperidol (H), haloperidol plus MDMA (HM), ketanserin (K), and ketanserin plus MDMA (KM). MDMA significantly increased PPI in the citalopram and the haloperidol group (*p < .05). Citalopram pretreatment significantly reduced the MDMA-induced increase in PPI (two-way ANOVA, pretreatment × treatment interaction). Neither haloperidol nor ketanserin reduced the increase in PPI produced by MDMA. The increase in PPI after ketanserin plus MDMA approached statistical significance (p < .06)
Startle Measures, Haloperidol, and MDMA
Startle magnitudes for the first and last block are shown in Figure 3. Habituation was confirmed for all drug conditions by three-way ANOVA with MDMA, haloperidol, and block as factors which revealed a significant main effect of block [F(2.2,24) = 45.27; p < .001]. There were no differences in habituation between drug conditions. There was a significant main effect of MDMA [F(1,11) = 4.71; p < .05]. Haloperidol had no significant effect on startle magnitude or habituation, whether given alone or as a pretreatment to MDMA.
Mean %PPI values are shown in Figure 4. Three-way ANOVA with MDMA, haloperidol, and PPI condition yielded a significant main effect of PPI condition [F1.6,18) = 10.86; p < .001], confirming prepulse inhibition. There was a significant main effect of MDMA [F(1,11) = 8.42; p < .01], but no haloperidol × MDMA interaction, indicating that haloperidol pretreatment had no effect on MDMA-induced increases in PPI. There were no gender differences.
Startle Measures, Ketanserin, and MDMA
Magnitudes for the first and last block of the ketanserin MDMA study are shown in Figure 3. Habituation was confirmed by three-way ANOVA with MDMA, ketanserin, and block as factors which revealed a significant main effect of block [F(1.3,14) = 48.15; p < .001]. There were significant main effects of both MDMA [F(1,11) = 6.45; p < .03] and of ketanserin [F(1,11) = 5.6; p < .04], but no interaction of the two drugs. Ketanserin alone lowered startle magnitude in all blocks but had no such effect as a pretreatment to MDMA.
Mean %PPI values are shown in Figure 4. Three-way ANOVA with MDMA, ketanserin, and PPI condition revealed a significant main effect of PPI [F(1.4,15) = 30.52; p < .001], no significant main effects of drug, and no significant ketanserin × MDMA interaction.
Although both MDMA and ketanserin showed a trend to increase PPI compared to placebo these effects were not significant [respectively, F(1,11) = 2.5; p < 1.4; F(1,11) = 1.95; p < 1.9]. Coadministration of MDMA and ketanserin was more effective in increasing PPI than each drug alone and the main effect of drug (ketanserin-MDMA vs placebo) just failed to reach statistical significance [F(1,11) = 4.46; p < .06].
Correlations Between Differences in Psychological Ratings and Changes in %PPI
The psychological changes produced by MDMA in all three dimensions of the OAV questionnaire significantly correlated with MDMA-induced increases in %PPI. %PPI increases for the PP60 and PP240, which were mainly affected by MDMA, were best correlated. Changes in OB (oceanic boundlessness) scores positively correlated with changes in %PPI for PP60 and PP240 [respectively, r = 0.349; n = 43, p < .02 and r = 0.408; n = 43, p < .01]. Changes in AED (anxious ego-dissolution) scores correlated with changes in %PPI for PP60 [r = 0.455; n = 43, p < .01]. Changes in VR (visionary restructuralization) scores also correlated changes in %PPI for PP60 and PP240 [respectively, r = 0.419; n = 43, p < .01 and r = 0.353; n = 43, p < .02]. Correlations were similar when calculated for male subjects only, but mostly did not reach significant values in the small sample of women. Similar correlations or trends were confirmed for each of the three studies when calculated separately.
Correlations Between Differences in Psychological Ratings and Changes in %Habituation
MDMA-induced psychological changes significantly correlated with MDMA-induced reductions in %habituation. As seen in Figure 5 , changes in OB scores negatively correlated with changes in %habituation [r = −0.559; n = 43, p < .001]. Changes in VR scores also negatively correlated with changes in %habituation [r = −0.372; n = 43, p < .02]. Correlations were comparable for male and female subjects.

Correlation between MDMA-induced changes in OB (oceanic boundlessness) scores and MDMA-induced changes in percent habituation compared to placebo, n = 43. Subjects with high scores in OB consistent with a more intense MDMA experience exhibited disrupted percent habituation. Most subjects with low OB scores showed an increase in percent habituation compared with placebo. (RSp = Spearman's rank order correlation)
Discussion
MDMA (1.5 mg/kg PO) increased prepulse inhibition (PPI) of the acoustic startle reflex in humans compared to placebo. This effect of MDMA was attenuated significantly by pretreatment with the selective serotonin inhibitor citalopram (40 mg IV), but not by the D2 antagonist haloperidol or the 5-HT2A/C antagonist ketanserin.
This study confirms results from a recent human study from our laboratory with a slightly higher dose of MDMA (1.7 mg/kg) that similarly increased PPI in humans. The effect of MDMA on PPI in that study was significant for the 120-ms interval but not for the 30-ms interval (Vollenweider et al. 1999a). Consistent with the previous report, here we found no change in PPI for the 30-ms interval and an increase for the 60-, 120-, and 240-ms intervals.
At the dose used, the effects of MDMA on %PPI were moderate (mean of 10–14% absolute increase compared to placebo) but significant. In addition, we found a series of significant correlations between psychological changes produced by MDMA and the MDMA-induced increase in %PPI. Thus, subjects with a stronger overall MDMA experience also showed higher increases in %PPI.
In contrast to the present findings in humans, serotonin releasers such as MDMA, MDEA, AET, and fenfluramine all impair PPI of the startle reflex in rats or mice (Dulawa and Geyer 1996; Kehne et al. 1996; Mansbach et al. 1989; Martinez and Geyer 1997; Vollenweider et al. 1999a). Several methodological differences could contribute to the opposite effects of MDMA on PPI in rodents and humans. The dose of MDMA used in this and our previous human study was lower than the doses shown to disrupt PPI in animals (Vollenweider et al. 1999a). In addition, in humans, the eyeblink component of the startle response is measured and in animals a whole-body flinch is registered. Such differences, however, are common to virtually all comparisons between drug effects in rodents and humans. Given the many other similarities between rodent and human findings in studies of PPI, the specific differences in the effects of MDMA might be better explained by differences in effects of MDMA on brain serotonin systems.
In the present study, pretreatment with the highly selective serotonin uptake inhibitor citalopram attenuated the effect of MDMA on PPI as well as MDMA-induced psychological changes (Liechti et al. 2000a). Similarly, the effects of MDMA-like serotonin releasers, such as fenfluramine or AET, on PPI and startle habituation were prevented by serotonin uptake inhibitors in rodents (Kehne et al. 1992; Martinez and Geyer 1997). These findings are in line with a large number of preclinical studies suggesting that MDMA acts primarily by releasing serotonin via a carrier-mediated mechanism that is blocked by selective serotonin uptake inhibitors (Callaway et al. 1990; Gu and Azmitia 1993; Gudelsky and Nash 1996; Hekmatpanah and Peroutka 1990; Koch and Galloway 1997). Based on this evidence, it seems that carrier-mediated serotonin release also contributes to the effect of MDMA on PPI in humans. Citalopram is currently the most selective SSRI available. It binds competitively to the 5-HT uptake site with high affinity and exhibits very low affinity for other receptors (Hyttel et al. 1995; Milne and Goa 1991). By blocking reuptake of 5-HT, citalopram also slightly increases the availability of 5-HT in the synaptic cleft, although to a lesser extent than the potent 5-HT releaser MDMA. However, in analogy to the large number of similar preclinical investigations, the primary action of citalopram in the present study was that of an antagonist at the 5-HT uptake site.
Nonspecific adverse effects of citalopram, such as nausea in some subjects, might account for the reduction of some of the pleasurable MDMA effects. However, the side effects of citalopram cannot explain the reduction of the different psychological and physiological effects of MDMA seen in all subjects in this study. Therefore, the fact that the MDMA-induced increase in PPI was attenuated by citalopram seems to indicate a specific interaction of citalopram and MDMA at the 5-HT uptake site.
Unlike citalopram, both haloperidol and ketanserin failed to interact with PPI effects produced by MDMA, although both were effective in reducing specific psychological effects of MDMA. In the present study, the D2 dopamine receptor antagonist haloperidol nonsignificantly reduced PPI whether given alone or as a pretreatment to MDMA. A significant reduction of PPI by haloperidol has been reported previously (Abduljawad et al. 1998), but could not be replicated by the same investigators (Abduljawad et al. 1999). In accordance with our results, in rodents, haloperidol did not reduce the effects of MDMA on PPI (Padich et al. 1996). Hence, it appears that D2 dopamine receptor stimulation is not primarily involved in the action of MDMA on PPI. These findings also indicate that the effect of MDMA on PPI both in animals and in humans is different from that of the classical stimulant d-amphetamine and the dopamine agonist bromocriptine, which appear to disrupt PPI via D2 stimulation (Abduljawad et al. 1998; Hutchison and Swift 1999).
In the present study, ketanserin nonsignificantly increased PPI and was ineffective in reducing the MDMA-induced increase in PPI. This finding, however, is limited by the fact that the MDMA-induced increase in the ketanserin-MDMA study did not reach significance. In animals, 5-HT2A antagonists such as M100907 reduced the MDMA-induced disruption of PPI (Padich et al. 1996). The discrepancy between animal studies and our finding with regard to postsynaptic 5-HT2A receptors remains unclear. However, the present finding is consistent with the view that stimulation of 5-HT2 receptors is not responsible for the increase in PPI produced by MDMA in humans.
The present confirmation of the hypothesis that MDMA increases PPI in humans by acting as an indirect serotonin agonist raises the question as to which serotonin receptor mediates the effects of the serotonin released by MDMA. In rats, activation of 5-HT1A, 5-HT1B, or 5-HT2A receptors disrupts PPI (Rigdon and Weatherspoon 1992; Sipes and Geyer 1994). In the present investigation, there was no evidence for a role of 5-HT2A receptors. Therefore, the observed difference in the effects of MDMA on PPI in humans and rats may be due to differences in 5-HT1 receptor stimulation. Indeed, there is precedent for species-specific differences in the effects of 5-HT1A agonists on PPI. In contrast to their PPI-disruptive effects in rats, 5-HT1A agonists increase PPI in mice (Dulawa et al. 1997). In both rodent species, these opposite effects are blocked by selective 5-HT1A antagonists (Dulawa et al. 1998; Sipes and Geyer 1995). No studies of 5-HT1A agonist effects on PPI in humans have been reported, although the hallucinogen psilocybin, which has partial agonist activity at both 5-HT1A and 5-HT2A receptors, appears to increase PPI slightly in humans (Gouzoulis-Mayfrank et al. 1998). Thus, it is possible that species-specific differences in 5-HT1A-mediated effects of the serotonin released by MDMA contribute to the opposite effects of MDMA on PPI in rodents versus humans. Indeed, marked neuroanatomical differences in the distribution of 5-HT1A binding sites were observed between rats and humans suggesting that different functional consequences may be produced after administration of drugs that influence 5-HT1A receptors (Duncan et al. 1998).
As yet, there are only a few human studies assessing pharmacological effects on PPI in humans to correlate findings with animal evidence. For example, the dopamine releaser D-amphetamine attenuated PPI in humans (Hutchison and Swift 1999), thereby mimicking the effects of amphetamine on PPI in both rats and mice. Similarly, as predicted by the many similar animal studies, the D2-agonist bromocriptine disrupted PPI in humans and this effect was blocked by the D2-antagonist haloperidol (Abduljawad et al. 1998). In contrast, but in accordance with our findings, the hallucinogenic serotonergic agonist psilocybin has been shown to increase PPI in humans (Gouzoulis-Mayfrank et al. 1998), while hallucinogens that are agonists at 5-HT2A receptors consistently disrupted PPI in animals; for a review see (Geyer 1998). While it is premature to draw firm conclusions from this limited evidence, we might assume that dopaminergic effects on PPI are more consistent in animals and humans, while there are differences in the effects of serotonergic drugs which might represent differences within the serotonin system across species. Additional studies will have to focus on the effect of these monoamines on PPI in humans extending evidence from mechanistic studies in animals to humans.
In the present study, MDMA produced moderate agitation, arousal, and an increase in facial muscle tone; these MDMA effects might have contributed to changes in startle reactivity. In contrast, percent PPI has been shown to be largely independent of the level of arousal or of startle magnitude (Schwarzkopf et al. 1993). In addition, PPI shows high within-subject stability across time and thus is a reliable measure that is appropriate for test-retest designs (Abel et al. 1998; Cadenhead et al. 1999; Schwarzkopf et al. 1993). Accordingly, we found no order effects of any treatment on any dependent variable.
Although MDMA did not affect %habituation compared to placebo in between-group comparisons, we found several highly significant correlations between MDMA-induced psychological changes and the reduction in %habituation. Subjects experiencing stronger overall psychoactive effects of MDMA showed pronounced habituation deficits. This finding indicates that higher doses would be necessary to disrupt %habituation in humans, similar to the disrupting effect of MDMA-like drugs on %habituation in animals (Martinez and Geyer 1997). Deficits in habituation have commonly been described in schizophrenic patients and may reflect reduced selective attention (Braff et al. 1992; Geyer and Braff 1982). In the present study, MDMA mainly induced euphoric effects indicated by increases in positive and mania-like mood (OB) and alterations in perception, such as changes in the meaning of percepts and facilitated recollection, that correlated significantly with decreased %habituation. Although it is premature to draw firm conclusions from these preliminary findings, these findings may be related to MDMA-induced serotonergic and dopaminergic overactivity. Future studies using MDMA and other psychoactive drugs should extend these results and might provide insight in the nature of information processing as assessed by PPI and habituation.
In conclusion, this is one of the first mechanistic studies assessing drug effects on PPI of the acoustic startle reflex in humans. MDMA increased PPI and this effect was attenuated by the selective serotonin reuptake inhibitor citalopram, but not by the D2 antagonist haloperidol or the 5-HT2A/C antagonist ketanserin. These findings support the hypothesis that MDMA exerts its effects on PPI in humans by releasing serotonin via a mechanism involving the serotonin uptake carrier. The MDMA-induced increase in PPI in humans is opposite to its effect in rodents, confirming our previous finding. It remains unclear whether methodological differences or differences within the serotonergic system between rodents and humans, possibly involving 5-HT1 receptors, account for this discrepancy. The present study demonstrates the compelling need to study the pharmacology of PPI in humans in order to provide comparison with the neurochemical information from animal studies of PPI and to increase our understanding of the basis of deficient sensorimotor gating in psychiatric disorders.
References
Abduljawad KAJ, Langley RW, Bradshaw CM, Szabadi E . (1998): Effects of bromocriptine and haloperidol on prepulse inhibition of the acoustic startle response in man. J Psychopharmacol 12: 239–245
Abduljawad KAJ, Langley RW, Bradshaw CM, Szabadi E . (1999): Effects of bromocriptine and haloperidol on prepulse inhibition: Comparison of the acoustic startle eyeblink response and the N1/P2 auditory-evoked response in man. J Psychopharmacol 13: 3–9
Abel K, Waikar M, Pedro B, Hemsley D, Geyer MA . (1998): Repeated testing of prepulse inhibition and habituation of the startle reflex: A study in healthy human controls. J Psychopharmacol 12: 330–337
Baumann P, Larsen F . (1995): The pharmacokinetics of citalopram. Rev Contemp Pharmacother 6: 287–295
Baumann P, Nil R, Bertschy G, Jecker A, Brändli H, Morand J, Kasas A, Vuagniaux O, Ramseier F . (1998): A double-blind double-dummy study of citalopram comparing infusion versus oral administration. J Affect Disord 49: 203–210
Bolino F, Di Michele V, Di Cicco L, Manna V, Daneluzzo E, Casacchia M . (1994): Sensorimotor gating and habituation evoked by electrocutaneous stimulation in schizophrenia. Biol Psychiatry 36: 670–679
Braff DL, Grillon C, Geyer MA . (1992): Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry 49: 206–215
Cadenhead KS, Carasso BS, Swerdlow NR, Geyer MA . (1999): Prepulse inhibition and habituation of the startle response are stable neurobiological measures in a normal male population. Biol Psychiatry 45: 360–364
Cadenhead KS, Geyer MA, Braff DL . (1993): Impaired startle prepulse inhibition and habituation in patients with schizotypal personality disorder. Am J Psychiatry 150: 1862–1869
Callaway CW, Wing LL, Geyer MA . (1990): Serotonin release contributes to the locomotor stimulant effects of 3,4-methylenedioxymethamphetamine in rats. J Pharmac Exp Ther 254: 456–464
Dittrich A . (1998): The standardized psychometric assessment of altered states of consciousness (ASCs) in humans. Pharmacopsychiat 31: 80–84
Dittrich A, von Arx S, Staub S . (1985): International study on altered states of consciousness (ISASC). Summary of the results. Germ J Psychol 9: 319–339
Dulawa SC, Geyer MA . (1996): Psychopharmacology of prepulse inhibition in mice. Chin J Physiol 39: 139–146
Dulawa SC, Hen R, Scearce-Levie K, Geyer MA . (1997): Serotonin 1B receptor modulation of startle reactivity, habituation, and prepulse inhibition in wild-type and serotonin 1B receptor knockout mice. Psychopharmacology 132: 125–134
Dulawa SC, Hen R, Scearce-Levie K, Geyer MA . (1998): 5-HT1B receptor modulation of prepulse inhibition: Recent findings in wild-type and 5-HT1B knockout mice. Ann NY Acad Sci 861: 79–84
Duncan GE, Knapp DJ, Breese GR, Crews FT, Little KY . (1998): Species differences in regional patterns of 3H-8-OH-DPAT and 3H-zolpidem binding in the rat and human brain. Pharmacol Biochem Behav 60: 439–448
Froemming JS, Francis Lam YW, Jann MW, Davis CM . (1989): Pharmacokinetics of haloperidol. Clin Pharmacokinet 17: 396–423
Geyer MA . (1998): Behavioral studies of hallucinogenic drugs in animals: Implications for schizophrenia research. Pharmacopsychiat 31: 73–79
Geyer MA, Braff DL . (1982): Habituation of the blink reflex in normals and schizophrenic patients. Psychophysiol 19: 1–6
Geyer MA, Callaway CW . (1994): Behavioral pharmacology of ring-substituted amphetamine analogs In Cho AK, Segal DS (eds), Amphetamine and Its Analogs: Psychopharmacology, Toxicology, and Abuse. San Diego, CA, Academic Press, 177–208
Gouzoulis-Mayfrank E, Heekeren K, Thelen B, Lindenblatt H, Kovar KA, Sass H, Geyer MA . (1998): Effects of the hallucinogen psilocybin on habituation and prepulse inhibition of the startle reflex in humans. Behav Pharmacol 9: 561–566
Gu XF, Azmitia EC . (1993): Integrative transporter-mediated release from cytoplasmic and vesicular 5-hydroxytryptamine stores in cultured neurons. Eur J Pharmacol 235: 51–57
Gudelsky GA, Nash JF . (1996): Carrier-mediated release of serotonin by 3,4-methylenedioxymethamphetamine: Implications for serotonin-dopamine interactions. J Neurochem 66: 243–249
Hekmatpanah CR, Peroutka SJ . (1990): 5-Hydroxytryptamine uptake blockers attenuate the 5-hydroxytryptamine-releasing effect of 3,4-methylenedioxymethamphetamine and related agents. Eur J Pharmacol 177: 95–98
Hutchison KE, Swift R . (1999): Effect of d-amphetamine on prepulse inhibition of the startle reflex in humans. Psychopharmacology 143: 394–400
Hyttel J, Arnt J, Sanchez C . (1995): The pharmacology of citalopram. Rev Contemp Pharmacother 6: 271–285
Janke W, Debus G . (1978): Die Eigenschaftswörterliste (EWL-K) - Ein Verfahren zur Erfassung der Befindlichkeit. Göttingen, Hogrefe
Kehne JH, Padich RA, McCloskey TC, Taylor VL, Schmidt CJ . (1996): 5-HT modulation of auditory and visual sensorimotor gating: I. Effects of 5-HTreleasers on sound and light prepulse inhibition in Wistar rats. Psychopharmacology 124: 95–106
Kehne JH, Timothy C, McCloskey TC, Taylor VL, Black CK, Fadayel GM, Schmidt CJ . (1992): Effects of the serotonin releasers 3,4-methylenedioxymethamphetamine (MDMA), 4-chloroamphetamine (PCA) and fenfluramine on acoustic and tactile startle reflexes in rats. J Pharmacol Exp Ther 260: 78–89
Koch S, Galloway MP . (1997): MDMA induced dopamine release in vivo: Role of endogenous serotonin. J Neural Transm 104: 135–146
Lieberman JA, Aghajanian GK . (1999): Caveat emptor: Researcher beware. Neuropsychopharmacology 21: 471–473
Liechti ME, Baumann C, Gamma A, Vollenweider FX . (2000a): Acute psychological effects of 3,4-methylenedioxymethamphetamine (MDMA, “Ecstasy”) are attenuated by the serotonin uptake inhibitor citalopram. Neuropsychopharmacology 22: 513–521
Liechti ME, Saur M, Gamma A, Hell D, Vollenweider FX . (2000a): Psychological and physiological effects of MDMA (“Ecstasy”) after pretreatment with the 5-HT2 antagonist ketanserin in healthy humans. Neuropsychopharmacology 23 (in press).
Liechti ME, Vollenweider FX . (2000a): The serotonin uptake inhibitor citalopram reduces acute cardiovascular and vegetative effects of MDMA (“Ecstasy”) in healthy volunteers. J Psychopharmacol 14: (in press).
Liechti ME, Vollenweider FX . (2000): Acute psychological and physiological effects of MDMA (”Ecstasy”) after haloperidol pretreatment in normal healthy humans. Eur Neuropsychopharmacol 10: 289–295b
Mansbach RS, Braff DL, Geyer MA . (1989): Prepulse inhibition of the acoustic startle response is disrupted by N-ethyl-3,4-methylenedioxyamphetamine (MDEA) in the rat. Eur J Pharmacol 167: 49–55
Mansbach RS, Geyer MA, Braff DL . (1988): Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology 94: 507–514
Martinez DL, Geyer MA . (1997): Characterization of the disruptions of prepulse inhibition and habituation of startle induced by a-Ethyltryptamine. Neuropsychophar- macology 16: 246–255
Milne RJ, Goa KL . (1991): Citalopram. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in depressive illness. Drugs 41: 450–477
Nordström A-L, Farde L, Halldin C . (1992): Time course of D2-dopamine receptor occupancy examined by PET after single oral doses of haloperidol. Psychopharmacology 106: 433–438
Padich RA, McCloskey TC, Kehne JH . (1996): 5-HT modulation of auditory and visual sensorimotor gating:II. Effects of the 5-HT2A antagonist MDL 100,907 on disruption of sound and light prepulse inhibition produced by 5-HT agonists in Wistar rats. Psychopharmacology 124: 107–116
Rigdon C, Weatherspoon J . (1992): 5-HT1A agonists block prepulse inhibition of the acoustic startle reflex. J Pharmacol Exp Ther 263: 486–493
Schwarzkopf SB, McCoy L, Smith DA, Boutros NN . (1993): Test-retest reliability of prepulse inhibition of the acoustic startle response. Biol Psychiatry 34: 896–900
Seifritz E, Baumann P, Müller MJ, Annen O, Amey M, Hemmeter U, Hatzinger M, Chardon F, Holsboer-Trachsler E . (1996): Neuroendocrine effects of a 20-mg citalopram infusion in healthy males. Neuropsychopharmacology 14: 253–263
Sills TL . (1999): Amphetamine dose dependently disrupts prepulse inhibition of the acoustic startle response in rats within a narrow time window. Brain Res Bull 48: 445–448
Sipes TA, Geyer MA . (1994): Multiple serotonin receptor subtypes modulate prepulse inhibition of the startle response in rats. Neuropharmacology 33: 441–448
Sipes TA, Geyer MA . (1995): 8-OH-DPAT disruption of prepulse inhibition in rats: Reversal with (+)WAY 100,135 and localization of site of action. Psychopharmacology 117: 41–48
Sipes TE, Geyer MA . (1997): DOI disrupts prepulse inhibition of startle in rats via 5-HT2A receptors in the ventral pallidum. Brain Res 761: 97–104
Swerdlow NR, Benbow CH, Zisook S, Geyer MA, Braff DL . (1993): A preliminary assessment of sensorimotor gating in patients with obsessive compulsive disorder (OCD). Biol Psychiatry 33: 298–301
Swerdlow NR, Hartman PL, Auerbach PP . (1996): Changes in sensorimotor inhibition across the menstrual cycle: Implications for neuropsychiatric disorders. Biol Psychiatry 41: 452–460
Swerdlow NR, Mansbach RS, Geyer MA, Pulvirenti L, Koob GF, Braff DL . (1990): Ampetamine disruption of prepulse inhibition of acoustic startle is reversed by depletion of mesolimbic dopamine. Psychopharmacology 100: 413–416
Swerdlow NR, Paulsen J, Braff DL, Butters N, Geyer MA, Swenson MR . (1995): Impaired prepulse inhibition of acoustic and tactile startle response in patients with Huntington's Disease. J Neurol Neurosurg Psychiat 58: 192–200
Vollenweider FX, Gamma A, Liechti ME, Huber T . (1998a): Psychological and cardiovascular effects and short-term sequelae of MDMA (“Ecstasy”) in MDMA-naive healthy volunteers. Neuropsychopharmacology 19: 241–251
Vollenweider FX, Vollenweider-Scherpenhuyzen MFI, Bäbler A, Vogel H, Hell D . (1998b): Psilocybin induces schizophrenia-like psychosis in humans via a serotonin-2 agonist action. NeuroReport 9: 3897–3902
Vollenweider FX, Remensberger S, Hell D, Geyer MA . (1999a): Opposite effects of 3,4-methylenedioxymethamphetamine (MDMA) on sensorimotor gating in rats versus healthy humans. Psychopharmacology 143: 365–372
Vollenweider FX, Gamma A, Liechti ME, Huber T . (1999b): Is a single dose MDMA harmless? Neuropsychopharmacology 21: 598–600
von Zerssen. D . (1976): Manual zur Beschwerden-Liste. Beltz, Weinheim
Acknowledgements
This study was supported by the Heffter Research Institute, Santa Fe, NM, USA. M. A. Geyer holds an equity interest in San Diego Instruments. The authors especially thank Alex Gamma, Ph.D., for critical comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liechti, M., Geyer, M., Hell, D. et al. Effects of MDMA (Ecstasy) on Prepulse Inhibition and Habituation of Startle in Humans after Pretreatment with Citalopram, Haloperidol, or Ketanserin. Neuropsychopharmacol 24, 240–252 (2001). https://doi.org/10.1016/S0893-133X(00)00199-8
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1016/S0893-133X(00)00199-8
Keywords
This article is cited by
-
Drug-drug interactions between psychiatric medications and MDMA or psilocybin: a systematic review
Psychopharmacology (2022)
-
Evaluation of drug incorporation into hair segments and nails by enantiomeric analysis following controlled single MDMA intakes
Analytical and Bioanalytical Chemistry (2016)
-
Changes in serotonin transporter (5-HTT) gene expression in peripheral blood cells after MDMA intake
Psychopharmacology (2015)
-
MDMA effects consistent across laboratories
Psychopharmacology (2014)
-
Human pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, ecstasy) after repeated doses taken 2 h apart
Psychopharmacology (2013)
