
Abstract
Some of the major concerns related to methamphetamine (METH) abuse are the neuronal damage inflicted at dopamine (DA) nerve terminals and the cognitive deficits observed in human METH abusers. We have shown that a high dose of METH selectively depleted dopaminergic markers in striatum, frontal cortex and amygdala of Swiss Webster mice, and impaired learned place preference. In this study, we investigated whether deficits in consolidation of place learning, as a consequence of METH neurotoxicity, underlie the underperformance of cocaine conditioned place preference (CPP). Administration of METH (5 mg/kg × 3) to Swiss Webster mice decreased striatal tyrosine hydroxylase (TH) immunoreactive neurons and significantly increased glial fibrillary acidic protein (GFAP) expression, confirming the neurotoxic potential of METH in mice. This treatment significantly attenuated the establishment of cocaine (15 mg/kg) CPP compared to control. To investigate whether manipulation of the consolidation phase improves learned place preference, mice were trained by cocaine and received daily post-training injections of DA receptor agonists or N-acetylcysteine (NAC). As memory consolidation occurs shortly after training, drugs were administered either immediately or 2 h post-training. Immediate post-training administration of the D1 DA receptor agonist SKF38393 (5, 10, and 20 mg/kg) or the D2 DA receptor agonist quinpirole (0.25, 0.5, and 1.0 mg/kg) did not improve the establishment of CPP following METH neurotoxicity. However, immediate but not delayed NAC administration (50 and 100 mg/kg) enhanced cocaine CPP following METH neurotoxicity and had no effect on control CPP. The levels of the reduced form of glutathione (GSH) in striatum, amygdala, hippocampus and frontal cortex were significantly lower in METH-treated mice compared to control during the period of CPP training. Acute and repeated administration of NAC to METH-treated mice restored the decreased brain GSH but had no effect on controls. Results suggest that METH-induced dopaminergic neurotoxicity is associated with impairment of consolidation of learned place preference, and that this impairment is improved by immediate post-training administration of the glutathione precursor NAC and not by D1 or D2 DA receptor agonists. Restoration of brain glutathione levels immediately post-training may facilitate the consolidation process.
Similar content being viewed by others


INTRODUCTION
Methamphetamine (METH) is a synthetic psychostimulant that in recent years has become a major drug of abuse. Evidence from animal and human studies suggests that long-term exposure or high dosages of amphetamines causes neuronal damage. Anatomical evidence supporting the neurotoxic potential of METH stems from studies showing that a high dose of METH given to rats caused degeneration of dopamine (DA) nerve terminals, and loss of tyrosine hydroxylase (TH), DA transporter (DAT), tryptophan hydroxylase (TPH), and 5-hydroxytryptamine (5-HT) axon terminals (Axt and Molliver, 1991; Gibb et al, 1990; Ricaurte et al, 1982; Seiden and Sabol, 1996). Further support for the neurotoxic potential of METH comes from evidence that (a) high doses of METH cause striatal gliosis as shown by an increase of glial fibrillary acidic protein (GFAP) in rats and mice (Battaglia et al, 2002; O'Callaghan and Miller, 1994; Sheng et al, 1994), and (b) apoptotic pathways are involved in METH-induced neuronal injury (Cadet et al, 2003).
Species-dependent differences in the neurotoxic effect of METH have been reported. METH causes concurrent dopaminergic and serotonergic neurotoxicity in rats (Lyles and Cadet, 2003), but rather selective dopaminergic neurotoxicity in nonhuman primates (Villemagne et al, 1998). In human METH abusers, selective reduction in the density of caudate and putamen DAT-binding sites has been associated with cognitive deficits (Chang et al, 2002; McCann et al, 1998; Volkow et al, 2001a, 2001b). In a series of previous studies, we found that high doses of METH in mice cause selective dopaminergic neurotoxicity with no evidence of serotonergic neurotoxicity (Itzhak and Achat-Mendes, 2004; Itzhak and Ali, 2006; reviews). Recently, we reported that METH-induced dopaminergic neurotoxicity in Swiss Webster mice selectively impaired place preference learning (Achat-Mendes et al, 2005).
The conditioned place preference (CPP) paradigm involves Pavlovian learning. Pairing of unconditioned stimulus (US; eg, reward) with a neutral context to become conditioned stimulus (CS) elicits approach behavior known as conditioned response (CR). The acquisition of place conditioning requires (a) US that changes the affective state of the organism and (b) learning and memory (White and Carr, 1985). The three major phases in Pavlovian conditioning include acquisition, consolidation and retrieval. The deficits in the development of cocaine CPP in mice pre-exposed to a neurotoxic regimen of METH, we observed (Achat-Mendes et al, 2005), could be the result of dysregulation of the DA-dependent affective state and impairment of associative learning. Lessening in cocaine reward (compared to control mice) may be the result of a decrease in DAT-binding sites, and impairment in associative learning may be due to the consequences of oxidative stress and DA depletion caused by METH. To what extent the decrease in DAT may weaken the affective state produced by cocaine is unclear. First, DAT knockout mice did not show deficits in cocaine CPP (Sora et al, 1998). Second, our finding that the bell-shaped dose–response of CPP was persistently reduced following METH-induced dopaminergic neurotoxicity compared to control suggests that ‘increasing reward’ does not improve CPP (Achat-Mendes et al, 2005). Third, the same neurotoxic treatment with METH induced psychomotor sensitization to cocaine, suggesting that diminished DAT-binding sites does not impede cocaine effects (Itzhak et al, 1997). Thus, the present study was undertaken to investigate whether manipulation of the consolidation phase of place learning can improve the deficits in CPP.
DA has a major role in appetitive conditioning by drug (Di Chiara, 2002) and natural (Phillips et al, 2003; Parkinson et al, 2002) reward. The findings that DA is also involved in aversive conditioning by LiCl (Di Chiara, 2002) and fear conditioning (Nader and LeDoux, 1999; Pezze and Feldon, 2004) support its role in associative learning and memory consolidation. Blockade of D1-like and D2-like DA receptors interferes with consolidation of Pavlovian conditioned behavior (Greba et al, 2001; Koch et al, 2000; LaLumiere et al, 2005; Nader and LeDoux, 1999). Given that METH neurotoxicity diminishes levels of corticostriatal and mesolimbic DA, we first sought to investigate if post-training administration of D1 and D2 DA receptor agonist improves consolidation of cocaine CPP.
Evidence suggests that oxidative stress and dopaminergic neurotoxicity are associated with depletion of brain glutathione and cognitive impairments (Banaclocha, 2001; Cruz et al, 2003; Dringen, 2000; Mandel et al, 2003; Weber, 1999). N-Acetylcysteine (NAC) is a pro-drug for the synthesis of the endogenous antioxidant glutathione, and was found to improve age-dependent memory impairments in mice (Farr et al, 2003; Martinez et al, 2000). We hypothesized that deficits in consolidation of CPP, subsequent to METH neurotoxicity, may be due in part to depletion of brain glutathione. Therefore, the effect of post-training administration of NAC on cocaine-induced CPP following METH neurotoxicity was also investigated. Results show that NAC but not D1 or D2 DA receptor agonists improved CPP consolidation following METH-induced dopaminergic neurotoxicity; the improvement may be due in part to the restoration of brain glutathione levels.
MATERIALS AND METHODS
Animals
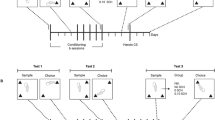
Male Swiss Webster mice (9 weeks old; 30–34 g; Charles River, Wilmington, MA) were maintained on a 12-h light/dark schedule at a room temperature of 22±0.5°C and housed in groups of five with free access to food and water. Animals were habituated for 5 days in the Division of Veterinary Resources (University of Miami School of Medicine) before drug treatments and then housed in groups of two following METH and saline administration. Animal care was in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, National Academy Press, 1996) and was approved by the University of Miami Animal Care and Use Committee. All drug treatments described below and saline were administered intraperitoneally (i.p.) in a volume of 0.1 ml/10 g body weight.
Pretraining Dopaminergic Neurotoxicity
Mice received saline or METH (5 mg/kg × 3; 3 h apart) 3 days before initiation of behavioral experiments. We have previously shown that METH (5 mg/kg × 3; 3 h apart) resulted in 42–61% depletion of DA- and DAT-binding sites in striatum, frontal cortex and amygdala of Swiss Webster mice (Achat-Mendes et al, 2005). Selective dopaminergic neurotoxicity with no evidence of serotonergic neurotoxicity (Itzhak and Achat-Mendes, 2004) was sustained for 95 days following METH administration to Swiss Webster mice (Itzhak et al, 2002). In the present study, METH-induced dopaminergic neurotoxicity was confirmed by immunohistochemical studies.
Immunohistochemistry
Mice received saline (n=3) or METH (5 mg/kg × 3; n=3) and after 5 days were anesthetized with a cocktail of ketamine (100 mg/kg) and xylazine (10 mg/kg). After loss of the foot-pinch response, mice were perfused via the left ventricle with sodium phosphate-buffered saline (PBS) followed by p-formaldehyde (4%) in PBS. The brain was removed and postfixed overnight in the same fixative at 4°C. Coronal sections (50 μm) were cut with a Vibratome, collected in PBS and blocked overnight (4°C) with normal goat serum (10%) in PB (0.1 M) containing Triton X-100 (0.3%). For double labeling, sections were incubated with a mouse monoclonal antibody to tryptophan hydroxylase (TH; 1:200; Chemicon) and rabbit polyclonal antibody to glial fibrillary acidic protein (GFAP; 1:200; Dako) (72 h; 4°C), diluted in a vehicle of normal goat serum (2%) with Triton X-100 (0.03%) in PB (0.1 M). Sections were rinsed 2 × 10 min in PBS and incubated (25°C; 30 min) with corresponding secondary antibodies: Alexa Fluor® 594 donkey anti-mouse IgG (TH) and Alexa Fluor® 488 goat anti-rabbit IgG (GFAP). Sections were rinsed in PBS (2 × 10 min), mounted onto slides, and coverslipped with Gel/Mount containing anti-fade agents. Negative control sections were treated in the same manner, except that the primary antibodies were omitted. Sections were analyzed by a fluorescence compound microscope (Olympus BX51; C. Squared Co., USA) equipped with a cooled monochrome camera (Retiga 2000R) and Image Pro Plus software.
Conditioning Apparatus
Place preference was monitored by a conditioning apparatus, the Opto-Max Activity Meter (Columbus Instruments, Columbus, OH). Each CPP cage (Plexiglas; 42 × 20 × 20 cm) was separated by a removable guillotine door into two compartments, one comprising four black walls and a smooth black floor and the other four white walls and a floor covered with beige sandpaper (fine grit 150C, Norton). Thus, the two compartments had different visual and tactile cues. During the habituation period and subsequently during testing of CPP the guillotine door was removed; during each training session the guillotine door was in place. The apparatus was covered with a transparent Plexiglas lid perforated to allow adequate ventilation. The cage was equipped with matching pairs of horizontal sensors mounted alongside opposing lengths (42 cm long). The black and white compartments (21 × 20 × 20 cm) were each scanned at a rate of 10 Hz by seven infrared beams, spaced at 2.54 cm intervals. A null zone 8 cm wide was assigned at the interface of the black and white zones and was monitored by two beams to ensure that only full entry into each compartment was registered as real time spent in each compartment. Information collected from sensors, for example, time spent in each compartment and horizontal locomotor activity was recorded and analyzed by the Opto-Max computer interface and v2.25-A software.
Conditioned Place Preference Training
Conditioned place preference (CPP) experiments commenced 3 days following the administration of METH and saline as we previously described (Achat-Mendes et al, 2005). The number of subjects in all CPP experiments described below was n=8–10. First, the habituation phase lasted for 2 days; mice were allowed free exploration of the black and white compartments of the CPP apparatus for 20 min; time spent in each compartment was recorded. Mice spent in average 485±41 s in the black and white compartments and 225±29 s in the null zone. Accordingly, mice were conditioned by cocaine in an unbiased manner: in each experiment half of the subjects were paired with drug in the black compartment and half in the white compartment. Secondly, the training phase consisted of eight sessions (once a day; 30 min each) of alternating pairings of cocaine (15 mg/kg) and saline (0.1 ml/10 g) injections (i.p.). During these sessions, a guillotine door with black and white walls was placed at the center of the CPP cage in order to separate the two compartments. At 1 day following the training, CPP was determined in a drug free state. Subjects were allowed free access to both compartments of the cage, and time spent in each compartment as well as locomotor activity was recorded for 20 min.
Posttraining Treatments
Experiment 1
The aim of the first experiment was to investigate if the attenuation of cocaine CPP following exposure to a neurotoxic regimen of METH can be ameliorated by posttraining administration of D1 or D2 DA receptors agonists or NAC. Mice received pretraining injections of METH (5 mg/kg × 3), and then were trained by cocaine (15 mg/kg) as described above. Immediately after each training session (cocaine or saline) mice received one of the following treatments: the D1 DA receptor agonist SKF38393 (5, 10 and 20 mg/kg; i.p.), the D2 DA receptor agonist quinpirole (0.25, 0.5 and 1.0 mg/kg; i.p.) or NAC (50 and 100 mg/kg; i.p.), after which they were returned to their home cage. Three control groups received the following: (1) pretraining saline, training by saline, and immediate post-training saline, (2) pretraining saline, training by cocaine, and immediate post-training saline, (3) pretraining METH, training by cocaine, and immediate post-training saline.
To investigate if post-training NAC administration improved recovery of DAT binding sites in METH-treated mice, the density of striatal DAT-binding sites was determined 24 h following termination of CPP testing. Saturation binding assays using [3H]mazindol in striatal tissue of saline/saline/saline, saline/cocaine/saline, METH/cocaine/saline, and METH/cocaine/NAC groups were carried out as previously described (Achat-Mendes et al, 2005).
Experiment 2
The second experiment was designed to further investigate the effects of post-training administration of NAC on cocaine CPP. The effects of immediate post-training NAC (100 mg/kg) administration on CPP following: (1) pretraining saline and training by saline, (2) pretraining saline and training by cocaine, and (3) pretraining METH and training by cocaine were investigated. Group 1 determined if NAC had affective properties that might influence subsequent place preference. Group 2 resolved if NAC influences cocaine CPP in control subjects. Group 3 confirmed the effect of NAC on cocaine CPP following METH neurotoxicity (as in Experiment 1). An additional METH group trained by cocaine received NAC (100 mg/kg) 2 h post-training. This group served as a control to determine whether NAC had a specific effect on memory consolidation; delayed administration of cognitive enhancing agents does not improve memory consolidation. Additional control groups were: (a) pretraining saline, training by cocaine and immediate post-training saline and (b) pretraining METH, training by cocaine and immediate post-training saline.
Experiment 3
The third experiment investigated if NAC facilitates CPP in control and METH pretreated mice that received suboptimal training, that is, four training sessions instead of eight. Five groups were investigated: (1) pretraining saline, training by cocaine and immediate post-training saline, (2) pretraining saline, training by cocaine, and immediate post-training NAC (100 mg/kg), (3) pretraining METH, training by cocaine, and immediate post-training saline, (4) pretraining METH, training by cocaine, and immediate post-training NAC (100 mg/kg), and (5) pretraining METH, training by cocaine and delayed post-training NAC (100 mg/kg). Subjects from each group were tested twice for CPP expression in a drug free state, after the first block of four training sessions and again after the second block of four training sessions.
Brain Glutathione Levels
The effects of METH and NAC on tissue content of glutathione were investigated in experiments, which were designed to simulate the time course of the CPP training. There were three control groups (n=4/group): (1) saline/saline, (2) saline/acute NAC (100 mg/kg); these were killed after 1 h, and (3) saline and chronic NAC (100 mg/kg × 8 days); this group was killed after 24 h. There were four METH (5 mg/kg × 3) groups (n=4/group): (1) acute saline 5 days after METH; killed after 1 h, (2) acute NAC (100 mg/kg) 5 days after METH; killed after 1 h, (3) repeated saline from days 5–12; killed on day 13, (4) chronic NAC (100 mg/kg) from days 5–12; killed on day 13. Bilateral frontal cortex, striatum, amygdala and hippocampus were dissected and stored at −80°C. Tissue from individual mice was homogenized in 10 volumes of potassium phosphate buffer (125 mM; pH7.4) centrifuged (13 000g; 5 min; 4°C) and the supernatant was diluted in six volumes of the same buffer. Aliquots were incubated (10 min; 24°C) in a final volume of 150 μl in the absence or presence of GSSG reductase (0.2 U/ml), β-nicotinamide adenine dinucleotide phosphate (NADPH; 0.2 mM) and 5,5′-dithiobis-2-nitrobenzoic acid (DTNB; 0.14 mM). Standard curves were generated in the presence of GSSG reductase and known concentrations of GSSG (oxidized GSH). Tissue levels of GSH (in the absence of reductase) and total glutathione (in the presence of reductase) were determined spectrophotometrically (ELx800, Bio-Tek Instruments) at 405 nm. Protein concentrations were determined by a Bio-Rad kit.
Data Analysis
CPP results are presented as the mean±SEM difference between the times spent in the drug-paired compartment before and after training. The effects of various drugs on CPP expression were analyzed by one-way ANOVA followed by post hoc Newman–Keuls test. Comparisons between CPP expression following four and eight training sessions were analyzed by two-way ANOVA (treatment × time) and post hoc Bonferroni test. Comparisons between locomotor activity pre- and post-training were also analyzed by two-way ANOVA (treatment × time) and post hoc Bonferroni test. Results of glutathione levels in each brain region were analyzed by one-way ANOVA followed by post hoc Newman–Keuls test. A significance criterion of p<0.05 was used for all analyses.
RESULTS
METH-Induced Neurotoxicity: Immunohistochemistry of TH and GFAP
Results in Figure 1 depict the effects of METH and saline on TH and GFAP immunoreactivity in mouse striatum. At 5 days following METH administration (5 mg/kg × 3) the intensity of striatal TH-positive neurons was reduced by 50±5% compared to control. The intensity of GFAP was increased by 85±10% of control. These findings confirm the development of dopaminergic neurotoxicity following the administration of this particular regimen of METH to Swiss Webster mice. The same schedule of METH inflicted 42–61% depletion of DA- and DAT-binding sites in striatum, frontal cortex and amygdala of Swiss Webster mice 2 weeks following METH administration (Achat-Mendes et al, 2005).

Effect of METH (5 mg/kg × 3; i.p.) on tyrosine hydroxylase (TH)-positive neurons (red) and glial fibrillary acidic protein (GFAP)-positive astocytes (green) in coronal section of mouse striatum. Immunohistochemical studies were carried out 5 days after saline and METH administration to Swiss Webster mice. (a) Normal pattern of TH and GFAP in saline-pretreated mice. Note that the green band in (a) represents astrocytes lining the septum and lateral ventricle, as expected. (b) Reduction in TH-positive neurons and increase in GFAP expression in METH pretreated mice. The intensity of TH staining was reduced by approximately 50% and that of GFAP was increased by approximately 80% of control. Arrows indicate regional gliosis. Bar represents 500 μm. Magnification: × 20.
Effects of Post-Training Administration of D1 and D2 Receptor Agonists and NAC on Cocaine CPP
As the aim of this study was to investigate whether METH neurotoxicity is associated with impairment in CPP consolidation, all drugs tested were administered post-CPP training (immediately or after a delay of 2 h).
Experiment 1
The first experiment investigated if attenuation of cocaine CPP following exposure to a neurotoxic regimen of METH is ameliorated by post-training administration of D1 and D2 DA receptors agonists and NAC. Results in Figure 2 show that pretraining administration of METH significantly inhibited the magnitude of cocaine CPP compared to a control group that received saline prior to training by cocaine (p<0.05). The D1 DA receptor agonist SKF38393 (5, 10 and, 20 mg/kg), the D2 DA receptor agonist quinpirole (0.25, 0.5, and 1.0 mg/kg) and NAC (50 and 100 mg/kg) were administered to METH pretreated groups daily immediately following each training session (cocaine and saline). Results in Figure 2 were analyzed by one-way ANOVA and yielded overall significant differences between the groups F[10,76]=6.99; p<0.0001. Neither SKF38393 nor quinpirole at the doses tested had significant effect on the magnitude of CPP in METH pretreated mice compared to a group that received saline instead of the D1 or the D2 DA receptor agonists (Figure 2). However, NAC (50 and 100 mg/kg) significantly increased the magnitude of CPP in the METH pretreated groups compared to a group that received saline instead of NAC (p<0.05). The magnitude of CPP in the METH groups that received post-training injections of NAC was not significantly different than the magnitude of CPP in control mice. These findings suggest that NAC but not D1 or D2 DA receptor agonists improved CPP consolidation in the METH groups.

Effect of posttraining administration of SKF38393, quinpirole and NAC on cocaine-induced CPP. Mice (n=8–10 per group) received pretraining injections of saline or METH (5 mg/kg × 3; i.p.). After 3 days mice were habituated to the CPP cages for 2 days and thereafter were trained by saline or cocaine (15 mg/kg; i.p.) for 8 days. Immediately after each training session mice were administered saline, SKF38393 (5, 10, and 20 mg/kg; i.p.), quinpirole (0.25, 0.5 and 1.0 mg/kg; i.p.) or NAC (50 and 100 mg/kg: i.p.) and returned to their home cage. CPP was determined in a drug-free state 24 h after the last training session. Abbreviations used: S=saline, C=cocaine, M=METH, NAC=N-acetylcysteine. +p<0.05 all groups vs S/S/saline control; *p<0.05 all groups vs S/C/saline group.
Locomotor activity in a drug-free state before training, that is, during the second day of habituation to the CPP cages, and thereafter during CPP testing was simultaneously recorded for 20 min (Figure 3). Results of all 11 groups (first experiment) were analyzed by two-way ANOVA (treatment × time) indicated insignificant treatment effect F[10,144]=0.995; p=0.449 and insignificant time effect F[1,144]=2.847; p=0.0937. These results suggest that the differences in the expression of CPP observed (Figure 2) cannot be attributed to variations in motor behavior during the pre- and post-training phases.

Locomotor activity during pretraining habituation to the CPP cages and following post-training during CPP testing. Results represent mean±SEM horizontal activity counts registered for 20 min during the second habituation day and then again post-training during CPP testing in a drug-free state. The 11 groups (n=8–10 per group) correspond to the same groups as in Figure 2. Abbreviations used: S=saline, C=cocaine, M=METH, NAC=N-acetylcysteine. A two-way ANOVA (treatment × time) yielded insignificant treatment effect F[10, 144]=0.995; p=0.449 and insignificant time effect F[1,144]=2.847; p=0.0937.
To investigate if the enhancement of CPP consequent to repeated NAC administration to the METH group was associated with recovery of DAT-binding sites, the density of striatal DAT-binding sites was determined in four of the groups depicted in Figure 2: saline/saline/saline, saline/cocaine/saline, METH/cocaine/saline, and METH/cocaine/NAC (100 mg/kg). Results in Figure 4 indicate that repeated administration of NAC had no significant effect on METH-induced depletion of striatal DAT-binding sites.

Comparison between the densities of striatal DAT-binding sites in saline and METH-pretreated mice, and the effect of post-training administration of NAC. Striatal tissue from four groups: (1) saline/saline/saline, (2) saline/cocaine/saline, (3) METH/cocaine/saline, and (4) METH/cocaine/NAC (100 mg/kg) described in Figures 2 and 3 was processed for determination of the densities of DAT-bindings sites labeled by [3H]mazindol. One-way ANOVA showed significant differences between the four groups F[3,8]=52.90; p<0.0001. Newman–Keuls post hoc test showed that the densities of DAT sites in the METH/cocaine/saline and METH/cocaine/NAC (100 mg/kg) groups were the same and significantly lower than the groups pretreated with saline (*p<0.05).
Experiment 2
The second experiment further investigated the effects of post-training administration of NAC on cocaine CPP. The effects of immediate post-training NAC administration (100 mg/kg) on CPP following (1) pretraining saline and training by saline, (2) pretraining saline and training by cocaine, and (3) pretraining METH and training by cocaine were investigated. An additional METH group trained by cocaine received NAC (100 mg/kg) 2 h post-training (delayed group; DEL in Figure 5). Results analyzed by one-way ANOVA yielded overall significant differences between the groups F[6,49]=13.82; p<0.0001. Post hoc analyses yielded the following: (1) an insignificant effect of post-training NAC administration on the control saline/saline group, suggesting that NAC had no affective properties which might influence subsequent place preference; (2) a significant cocaine training effect on the saline group (p<0.001) but insignificant post-training NAC effect (p>0.05), indicating that NAC had no effect on CPP magnitude in the control group; (3) a significant pretraining METH effect on CPP magnitude compared to control (p<0.05) and a significant post-training effect of NAC on CPP magnitude (p<0.05), a finding that confirms the results in Figure 2; (4) an insignificant effect of delayed NAC administration in the METH group. The magnitude of CPP in the ‘delayed’ group was significantly lower than the magnitude of CPP in the ‘immediate’ group (p<0.05). This finding suggests that the enhancement of CPP was due to improvement of CPP consolidation.

Effect of immediate and delayed post-training administration of NAC on cocaine CPP. Mice (n=8 per group) received pretraining injections of saline or METH (5 mg/kg × 3; i.p.). After 3 days mice were habituated to the CPP cages for 2 days and thereafter were trained by saline or cocaine (15 mg/kg: i.p.) for 8 days. Immediately after each training session mice were administered saline or NAC (100 mg/kg; i.p.) upon which they were returned to home cage. The specific effect of NAC on CPP consolidation was determined by a negative control group (M/C/NAC) that received delayed (2 h) post-training injection of NAC. Abbreviations used: S=saline, C=cocaine, M=METH, NAC=N-acetylcysteine, IMM=immediate, DEL=delayed. +p<0.05 all groups vs S/S/S IMM control; *p<0.05 M/C/S IMM vs S/C/S IMM and M/C/NAC DEL vs M/C/NAC IMM. Note that delayed administration of NAC had no effect compared to immediate administration of NAC to the M/C groups. Immediate NAC administration had no significant effect on S/S and S/C control groups.
Experiment 3
As the lack of effect of NAC on CPP magnitude in control mice may be due to a ‘ceiling effect,’ the third experiment investigated if NAC facilitates CPP in control and METH pretreated mice that received suboptimal training, that is, four training sessions instead of eight. Results in Figure 6 depicts the magnitude of CPP in control and METH pretreated mice following four and eight training sessions, and the effects of immediate and delayed post-training administration of NAC (100 mg/kg). Results were analyzed by two-way ANOVA (treatment × time): there was a significant overall treatment effect F[4,80]=5.72; p<0.001, a significant time effect F[1,80]=13.46; p<0.001 and marginal significant interaction F[4,80]=2.44; p=0.0534. Post hoc analyses revealed the following: (1) in control groups, eight training sessions resulted in significantly greater CPP than four training sessions (p<0.01). Post-training administration of NAC to the control group had no significant effect on CPP following four and eight training sessions; (2) in the METH group that received post-training saline injections, the magnitude of CPP after four and eight training sessions was the same, and lower than control (eight training sessions); (3) in the METH group that received immediate post-training NAC injections the magnitude of CPP following four and eight training sessions was significantly higher than CPP in the METH group that received saline instead of NAC (p<0.05); (4) in the METH group that received delayed posttraining NAC injections the magnitude of CPP following four and eight training sessions was the same, and insignificantly different from the METH group that received saline instead of NAC (p>0.05). The results suggest that NAC had selective effect on CPP consolidation in METH pretreated mice.

Effect of post-training NAC administration on cocaine CPP following four and eight training sessions. Mice (n=8 per group) received pretraining injections of saline or METH (5 mg/kg × 3; i.p.). After 3 days mice were habituated to the CPP cages for 2 days and thereafter were trained by saline or cocaine (15 mg/kg; i.p.). Immediately after each training session mice were administered saline or NAC (100 mg/kg; i.p.) upon which they were returned to home cage. To investigate if post-training NAC administration improves CPP consolidation in the course of suboptimal training, subjects were tested following four and eight training sessions. Immediate post-training NAC administration to control group trained by cocaine for 4 days (S/C/NAC IMM) did not improve CPP expression compared to the corresponding group that received saline (S/CS/IMM). In both groups, eight training sessions resulted in significantly larger CPP compared to four training sessions (*p<0.05). However, immediate post-training NAC administration to the METH group trained by cocaine for 4 and 8 days (M/C/NAC IMM) resulted in significantly larger CPP compared to corresponding group that received saline (M/C/S IMM) (+p<0.05). Delayed post-training NAC administration had no significant effect on the METH group trained by cocaine for 4 and 8 days. Two-way ANOVA yielded significant overall treatment effect F[4,80]=5.72; p<0.001, a significant time effect F[1,80]=13.46; p<0.001 and marginal significant interaction F[4,80]=2.44; p=0.0534. Abbreviations used: S=saline, C=cocaine, M=METH, NAC=N-acetylcysteine, IMM= immediate, DEL=delayed.
Effect of METH and NAC on Brain Glutathione Levels
The effects of METH and NAC on tissue content of GSH were investigated in experiments that were designed to simulate the time course of the CPP training. To determine the absolute level of GSH, assays were carried out in the absence of GSSG reductase, which reduces GSSG to GSH. Five and 13 days following METH administration (corresponding to the first and last days of CPP training) GSH concentration in the frontal cortex, striatum, amygdala, and hippocampus were reduced by 18–34% of control levels (p<0.05) (Table 1). At 1 h after acute administration of NAC (100 mg/kg) on the fifth day following METH administration, GSH levels were similar to control (Table 1). Following the chronic administration of NAC to the METH group (corresponding to the 8-day of CPP training; group ‘Day 13 NAC chronic’ Table 1) levels of GSH were insignificantly different from control. In a second series of experiments, assays were carried out in the presence of reductase (0.2 U/ml). Results showed that levels of total glutathione (GSH plus GSSG which had been converted to GSH) in frontal cortex, striatum, amygdala, and hippocampus of METH-treated mice were not significantly different from control (data not shown). This finding suggests that diminished levels of endogenous GSH may be due to increase in levels of endogenous GSSG, which were converted in vitro to GSH in the presence of reductase.
DISCUSSION
The major findings of the present study are that METH-induced dopaminergic neurotoxicity (a) impairs consolidation of learned place preference, which is ameliorated by systemic post-training administration of the glutathione precursor NAC and (b) reduces GSH concentrations in the frontal cortex, striatum, amygdala, and hippocampus, which are restored to control levels after NAC treatment. DA receptor agonists did not facilitate consolidation of place preference learning, indicating that METH-induced DA deficiency per se may not be the mechanism by which CPP was impaired.
Given the implication of dopaminergic deficits in cognitive impairments in METH abusers, the consequences of METH-induced selective dopaminergic neurotoxicity on place learning in the mouse model were investigated. Administration of METH (5 mg/kg × 3) to Swiss Webster mice resulted in 42–61% depletion of DA and DAT in the striatum, frontal cortex and amygdala (Achat-Mendes et al, 2005; Itzhak and Achat-Mendes, 2004). In the present study, dopaminergic neurotoxicity was confirmed by the marked reduction of striatal TH-immunoreactive neurons and the significant increase in GFAP expression 5 days after METH administration to Swiss Webster mice (Figure 1). This neurotoxic regimen of METH impaired the development of cocaine CPP (Achat-Mendes et al, 2005 and Figure 2). Several lines of evidence suggest that diminished DAT-binding sites per se consequent to METH neurotoxicity cannot explain the diminished cocaine CPP. First, DAT knockout mice have developed cocaine CPP to the same degree as wild-type mice (Sora et al, 1998). Second, partial depletion of striatal DAT-binding sites following administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or METH to Swiss Webster mice had either no effect on or increased the sensitivity to cocaine-induced hyperlocomotion, respectively. These findings suggest that reduction in DAT-binding sites does not impede cocaine effects (Itzhak et al, 1997). Third, post-training administration of NAC- to METH-pretreated mice restored the expression of cocaine CPP but not the level of striatal DAT-binding sites (Figure 4), suggesting that the improvement in CPP was independent of the density of DAT-binding sites. These findings imply that putative changes in DA-dependent affective state due to METH neurotoxicity are insufficient to explain the impairment in cocaine CPP.
Therefore, in the present study, we investigated if METH neurotoxicity is associated with deficits in the consolidation process of CPP. Given the affective properties of DA receptor agonists (Hoffman and Beninger, 1988; White et al, 1991), it is impractical to investigate the effects of these drugs on acquisition of CPP, that is, before training sessions. Hence, the effect of D1 and D2 DA receptor agonists on the consolidation phase of place learning was investigated. The results showed that post-training administration of either the D1 DA receptor agonist SKF38393 or the D2 DA receptor agonist quinpirole had no effect on consolidation of cocaine CPP following dopaminergic neurotoxicity. The doses of DA receptor agonist tested are comparable to the doses used to investigate improvement of memory consolidation in inhibitory avoidance task in mice (Costanzi et al, 2004; Mele et al, 1996).
The roles of DA receptor subtypes in the acquisition and consolidation of Pavlovian conditioning are ambiguous. Blockade of nucleus accumbens core D1 and NMDA receptors inhibited the acquisition of appetitive instrumental Pavlovian conditioning (Smith-Roe and Kelley, 2000) but had no effect on memory consolidation (Hernandez et al, 2005). Although CPP is noninstrumental Pavlovian conditioning, the finding that activation of D1 receptors had no effect on CPP consolidation may be in accord with the latter study. However, in other paradigms of noninstrumental appetitive Pavlovian conditioning, post-training blockade of nucleus accumbens D1 and NMDA receptors inhibited memory consolidation (Dalley et al, 2005). The complexity of DA involvement in memory consolidation is further illustrated by findings that post-training administration of the D2 DA receptor antagonist sulpiride into rat nucleus accumbens impaired (Setlow and McGaugh, 1998), while its systemic administration to rats enhanced (Setlow and McGaugh, 2000) memory of spatial water maze learning. The reasons for the apparent discrepancies are unclear but may be due to the different training procedures and temporal issues of drug administration.
An alternative mechanism by which METH could impair memory consolidation is via oxidative stress and consequent neurotoxicity (Mandel et al, 2003). Two major pathways have been demonstrated to enhance oxidative stress: (a) Generation of reactive oxygen species (ROS), which inhibits high affinity glutamate uptake and thereby increases extracellular glutamate and (b) Continuous exposure to glutamate, which inhibits cystine uptake via the glutamate-cystine exchange system and thereby reduces cellular glutathione re-synthesis, leading to oxidative stress and cell death (Weber, 1999). Reduced glutathione (GSH) is a tripeptide containing glutamate, cysteine, and glycine. GSH depletion is one of the hallmarks of a number of disorders associated with oxidative stress (Weber, 1999). The majority of GSH uptake and release occurs in astroglial cells, but cysteine and cysteine donors such as NAC are used by neurons to synthesize and release GSH (Dringen, 2000). High doses of METH increased striatal glutamate (Mark et al, 2004; Stephans and Yamamoto, 1994), which can activate either of the aforementioned pathways to result in oxidative stress and neurotoxicity. Additionally, increases in striatal, frontocortical, and hippocampal GSH were observed in both rats (Harold et al, 2000) and mice (Flora et al, 2002) shortly after METH administration; other studies in rats found a decrease in striatal GSH after METH administration (Acikgoz et al, 2001; Moszczynska et al, 1998). It is likely that GSH and glutathione peroxidase are elevated shortly after METH administration in response to defensive mechanisms against oxidative stress, but GSH is subsequently depleted. For instance, immediately following in vivo administration of the dopaminergic neurotoxin MPTP to rats a massive increase (5000%) of striatal GSH was observed (Han et al, 1999). However, it is well known that in Parkinson's disease there is reduction (40–50%) of GSH in substantia nigra and putamen (Dringen, 2000; Mandel et al, 2003). Likewise, in postmortem brains of METH abusers a marked loss of DA in the caudate was accompanied by a decrease in GSH and an increase in GSSG, the oxidized product of GSH (Mirecki et al, 2004).
Notably, several lines of evidence point to the role of GSH in dopaminergic neurotoxicity and in learning and memory. Depletion of GSH in PC12 cells increased DA toxicity and cell death that was prevented by addition of GSH or NAC (Offen et al, 1996). In vitro application of METH to human brain endothelial cells reduced total GSH levels and activated AP-1 and NFkB, which could lead to upregulation of inflammatory genes (Lee et al, 2001). In astroglial cells, DA could interact directly with GSH or indirectly by generation of hydrogen peroxide, which causes a decrease in extracellular GSH and an increase in GSSG (Hirrlinger et al, 2002). Furthermore, reduction in brain glutathione is associated with learning and memory impairments, and dysfunction of the glutathione cycle may be instrumental in conditions such as senile dementia, Huntington chorea and Parkinson's disease (Banaclocha, 2001; Weber, 1999). Chronic administration of NAC improved memory impairments in aged mice (Farr et al, 2003; Martinez et al, 2000).
Given the role of oxidative stress in METH-induced dopaminergic neurotoxicity, we hypothesized that impairment in cocaine CPP may be due, in part, to depletion of glutathione. Thus, the effect of post-training administration of NAC on CPP consolidation was investigated. Results indicate that 50 and 100 mg/kg NAC completely restored the magnitude of CPP in mice pre-exposed to a neurotoxic regimen of METH compared to controls. The effect of NAC was specific to the impaired consolidation of CPP since (a) immediate, but not delayed, post-training administration increased the expression of CPP, and (b) it was observed in METH pre-exposed subjects, but not in controls. The premise that NAC had no effect on CPP consolidation in control subjects is further supported by the finding that the drug had no effect on control mice that had been suboptimally trained by cocaine (4 days instead of 8; Figure 6).
Pharmacokinetic studies indicate the likelihood that NAC crosses the blood brain barrier (BBB). Mouse studies have shown that NAC completely crosses the capillary wall to enter brain tissue and extracellular space, and that the amount of NAC transported across the BBB is well within the therapeutic range of CNS-active agents (Farr et al, 2003). Also, an increase of plasma GSH should increase brain GSH since the tripeptide also crosses the BBB, possibly by Na+-dependent GSH transport (Kannan et al, 1990).
The results of glutathione assays revealed that the neurotoxic regimen of METH caused significant reductions in GSH in striatum, frontal cortex, hippocampus, and amygdala; these brain regions are involved in place preference learning. The reduction in GSH was observed 5 and 13 days after METH administration, a time course that corresponds to the first and last days of the CPP training. Thus, it is likely that brain GSH remained lower than control during the period of CPP training. Acute administration of NAC resulted in elevated brain GSH in METH, but not saline, pretreated mice, indicating that the effect is specific to METH-induced deficits and that NAC does not elevate brain glutathione under basal conditions. This observation is in accord with the lack of effect of NAC on CPP consolidation in control mice that underwent suboptimal training (four sessions instead of eight). The finding that 24 h after 8-day administration of NAC to the METH group, brain GSH levels reached control levels implies that either immediate or delayed NAC administration may restore GSH levels. However, the finding that only the immediate, but not the delayed, NAC administration improved CPP expression suggests that restoration of GSH levels alone may not be sufficient to explain the improvement in CPP. It is rather the point in time of NAC administration and the elevation of GSH after each training session that had a significant effect on the improvement of place preference learning.
In the present study, GSH depletion may be due to: (a) reduced activity of the enzymes involved in the synthesis of glutathione, (b) depletion of the precursors glutamate, cysteine, and glycine, or (c) oxidative stress that oxidized GSH to GSSG. The last prospect is supported by the finding that when assays were carried out in the presence of GSSG reductase, the levels of GSH in METH samples were similar to control. It is likely that excess GSSG in the METH samples was reduced in vitro to GSH and thus total glutathione levels were not significantly different from control (data not shown).
The mechanism by which NAC improved CPP consolidation in the present study is not entirely clear. The finding that repeated NAC administration to the METH group had no effect on the diminished density of striatal DAT-binding sites (Figure 4) suggests that NAC did not reverse the deficits in DAT. NAC had no affective properties, which could have modulated CPP expression, because it influenced neither the saline-pre-trained/saline-trained mice nor the saline-pre-trained/cocaine-trained mice (Figure 5). The findings that (a) METH-induced dopaminergic neurotoxicity was accompanied by a decrease in GSH levels in substrates involved in Pavlovian conditioning, and (b) administration of NAC restored the levels of GSH, suggest that the decrease in GSH levels may contribute in part to deficits in CPP consolidation. Given that immediate but not delayed post-training administration of NAC improved CPP expression, it appears that NAC/GSH is required during the consolidation phase of place learning and does not function as a general cognitive enhancer.
Another mechanism by which NAC could improve consolidation of cocaine CPP involves the effects of NAC/GSH on glutamatergic transmission. For instance, systemic administration of NAC to cocaine-withdrawn rats increased extracellular levels of glutamate in the nucleus accumbens (Baker et al, 2003) via the cystine-glutamate exchange system (Warr et al, 1999). In addition, evidence suggests the co-activation of ionotropic glutamate receptors through direct interaction with GSH (Oja et al, 2000; Varga et al, 1997). Hence, facilitation of glutamatergic transmission by NAC/GSH can improve memory consolidation. Further studies are required to determine whether modulation of glutamate transmission following dopaminergic neurotoxicity improves consolidation of place preference learning.
In summary, the present study demonstrates that dopaminergic neurotoxicity is associated with impairment of consolidation of learned place preference, and that this impairment is improved by systemic post-training administration of the glutathione precursor NAC, but not D1 and D2 DA receptor agonists. The deficits in consolidation observed may be due in part to a decrease in glutathione in hippocampus, amygdala, frontal cortex, and striatum, brain regions that have been implicated in place preference learning. Immediate post-training administration of NAC, which restored brain glutathione levels, improved CPP consolidation. Deficits in appetitive conditioning by drug reward following METH neurotoxicity may have implications for human METH abusers, for example, impairment in memory consolidation and perhaps dysregulation of affective state may in part perpetuate the escalation in drug use in order to ‘compensate’ for a subsequently weakened ‘drug-experience’. Further studies will be required to investigate if NAC can improve other cognitive deficits associated with METH abuse.
References
Achat-Mendes C, Ali SF, Itzhak Y (2005). Differential effects of amphetamines-induced neurotoxicity on appetitive and aversive Pavlovian conditioning in mice. Neuropsychopharmacology 30: 1129–1137.
Acikgoz O, Gonenc S, Gezer S, Kayatekin BM, Uysal N, Semin I et al (2001). Methamphetamine causes depletion of glutathione and an increase in oxidized glutathione in the rat striatum and prefrontal cortex. Neurotox Res 3: 277–280.
Axt KJ, Molliver ME (1991). Immunocytochemical evidence for methamphetamine-induced serotonergic axon loss in the rat brain. Synapse 9: 302–313.
Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S et al (2003). Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci 6: 743–749.
Banaclocha MM (2001). Therapeutic potential of N-acetylcysteine in age-related mitochondrial neurodegenerative diseases. Med Hypotheses 56: 472–477.
Battaglia G, Fornai F, Busceti CL, Aloisi G, Cerrito F, De Blasi A et al (2002). Selective blockade of mGlu5 metabotropic glutamate receptors is protective against methamphetamine neurotoxicity. J Neurosci 22: 2135–2141.
Cadet JL, Jayanthi S, Deng X (2003). Speed kills: cellular and molecular bases of methamphetamine-induced nerve terminal degeneration and neuronal apoptosis. FASEB J 17: 1775–1788.
Chang L, Ernst T, Speck O, Patel H, DeSilva M, Leonido-Yee M et al (2002). Perfusion MRI and computerized cognitive test abnormalities in abstinent methamphetamine users. Psychiatry Res 114: 65–79.
Costanzi M, Battaglia M, Rossi-Arnaud C, Cestari V, Castellano C (2004). Effects of anandamide and morphine combinations on memory consolidation in cd1 mice: involvement of dopaminergic mechanisms. Neurobiol Learn Memory 81: 144–149.
Cruz R, Almaguer MW, Bergado RJA (2003). Glutathione in cognitive function and neurodegeneration. Rev Neurol 36: 877–886.
Dalley JW, Laane K, Theobald DE, Armstrong HC, Corlett PR, Chudasama Y et al (2005). Time-limited modulation of appetitive Pavlovian memory by D1 and NMDA receptors in the nucleus accumbens. Proc Natl Acad Sci USA 102: 6189–6194.
Di Chiara G (2002). Nucleus accumbens shell and core dopamine: differential role in behavior and addiction. Behav Brain Res 137: 75–114.
Dringen R (2000). Metabolism and functions of glutathione in brain. Prog Neurobiol 62: 649–671.
Farr SA, Poon HF, Dogrukol-Ak D, Drake J, Banks WA, Eyerman E et al (2003). The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem 84: 1173–1183.
Flora G, Lee YW, Nath A, Maragos W, Hennig B, Toborek M (2002). Methamphetamine-induced TNF-alpha gene expression and activation of AP-1 in discrete regions of mouse brain. Neuromolecular Med 2: 71–85.
Gibb JW, Johnson M, Hanson GR (1990). Neurochemical basis of neurotoxicity. Neurotoxicol 11: 317–322.
Greba Q, Gifkins A, Kokkinidis L (2001). Inhibition of amygdaloid dopamine D2 receptors impairs emotional learning with fear-potentiated startle. Brain Res 899: 218–226.
Han J, Cheng FC, Yang Z, Dryhurst G (1999). Inhibitors of mitochondrial respiration, iron (II), and hydroxyl radical evoke release and extracellular hydrolysis of glutathione in rat striatum and substantia nigra: potential implications to Parkinson's disease. J Neurochem 73: 1683–1695.
Harold C, Wallace T, Friedman R, Gudelsky G, Yamamoto B (2000). Methamphetamine selectively alters brain glutathione. Eur J Pharmacol 400: 99–102.
Hernandez PJ, Andrzejewski ME, Sadeghian K, Panksepp JB, Kelley AE (2005). AMPA/kainite, NMDA, and dopamine D1 receptor function in the nucleus accumbens core: a context-limited role in the encoding and consolidation of instrumental memory. Learn Memory 12: 285–295.
Hirrlinger J, Schulz JB, Dringen R (2002). Effects of dopamine on the glutathione metabolism of cultured astroglial cells: implications for Parkinson's disease. J Neurochem 82: 458–467.
Hoffman DC, Beninger JR (1988). Selective D1 and D2 dopamine agonists produce opposing effects in place conditioning but not in conditioned taste aversion learning. Pharmacol Biochem Behav 31: 1–8.
Itzhak Y, Achat-Mendes C (2004). Methamphetamine and MDMA (ecstasy) neurotoxicity: ‘of mice and men’. IUBMB Life 56: 249–255.
Itzhak Y, Ali SF (2006). Role of nitrergic system in behavioral and neurotoxic effects of amphetamine analogs. Pharmacol Ther 109: 246–262.
Itzhak Y, Martin JL, Ali SF (2002). Methamphetamine-induced dopaminergic neurotoxicity in mice: long-lasting sensitization to the locomotor stimulation and desensitization to the rewarding effects of methamphetamine. Prog Neuropsychopharmacol Biol Psychiatry 26: 1177–1183.
Itzhak Y, Martin J, Ali SF, Norenberg MD (1997). Depletion of striatal dopamine transporter does not affect psychostimulant-induced locomotor activity. Neuro Report 8: 3245–3249.
Kannan R, Kuhlenkamp JF, Jeandidier E, Trinh H, Ookhtens M, Kaplowitz N (1990). Evidence for carrier-mediated transport of glutathione across the blood-brain barrier in the rat. J Clin Investig 85: 2009–2013.
Koch M, Schmid A, Schnitzler HU (2000). Role of nucleus accumbens dopamine D1 and D2 receptors in instrumental Pavlovian paradigm of conditioned reward. Psychopharmacol 152: 67–73.
LaLumiere RT, Nawar EM, McGaugh JL (2005). Modulation of memory consolidation by the basolateral amygdala or nucleus accumbens shell requires concurrent dopamine receptor activation in both brain regions. Learn Memory 12: 296–301.
Lee YW, Hennig B, Yao J, Toborek M (2001). Methamphetamine induces AP-1 and NF-kappaB binding and transactivation in human brain endothelial cells. J Neurosci Res 66: 583–591.
Lyles J, Cadet JL (2003). Methylenedioxymethamphetamine (MDMA, Ecstasy) neurotoxicity: cellular and molecular mechanisms. Brain Res Rev 42: 155–168.
Mandel S, Grunblatt E, Riederer P, Gerlach M, Levites Y, Youdim MB (2003). Neuroprotective strategies in Parkinson's disease: an update on progress. CNS Drugs 17: 729–762.
Mark KA, Soghomonian JJ, Yamamoto BK (2004). High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J Neurosci 24: 11449–11456.
Martinez M, Hernandez AI, Martinez N (2000). N-Acetylcysteine delays age-associated memory impairment in mice: role in synaptic mitochondria. Brain Res 855: 100–106.
McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA (1998). Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J Neurosci 18: 8417–8422.
Mele A, Castellano C, Felici A, Cabib S, Caccia S, Oliverio A (1996). Dopamine-N-methyl-D-aspartate interactions in the modulation of locomotor activity and memory consolidation in mice. Eur J Pharmacol 308: 1–12.
Mirecki A, Fitzmaurice P, Ang L, Kalasinsky KS, Peretti FJ, Aiken SS et al (2004). Brain antioxidant systems in human methamphetamine users. J Neurochem 89: 1396–1408.
Moszczynska A, Turenne S, Kish SJ (1998). Rat striatal levels of the antioxidant glutathione are decreased following binge administration of methamphetamine. Neurosci Lett 255: 49–52.
Nader K, LeDoux JE (1999). Inhibition of the mesoamygdala dopaminergic pathway impairs the retrieval of conditioned fear associations. Behav Neurosci 113: 891–901.
O'Callaghan JP, Miller DB (1994). Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther 270: 741–751.
Offen D, Ziv I, Sternin H, Melamed E, Hochman A (1996). Prevention of dopamine-induced cell death by thiol antioxidants: possible implications for treatment of Parkinson's disease. Exp Neurol 141: 32–39.
Oja SS, Janaky R, Varga V, Saransaari P (2000). Modulation of glutamate receptor functions by glutathione. Neurochem Intl 37: 299–306.
Parkinson JA, Dalley JW, Cardinal RN, Bamford A, Fehnert B, Lachenal G et al (2002). Nucleus accumbens dopamine depletion impairs both acquisi-tion and performance of appetitive Pavlovian approach behaviour: implications for meso-accumbens dopamine function. Behav Brain Res 137: 149–163.
Pezze MA, Feldon J (2004). Mesolimbic dopaminergic pathways in fear conditioning. Prog Neurobiol 74: 301–320.
Phillips GD, Setzu E, Hitchcott PK (2003). Facilitation of appetitive Pavlovian conditioning by d-amphetamine in the shell, but not the core, of the nucleus accumbens. Behav Neurosci 117: 675–684.
Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY (1982). Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res 235: 93–103.
Seiden LS, Sabol KE (1996). Methamphetamine and methylenedioxymethamphetamine neurotoxicity: possible mechanisms of cell destruction. NIDA Res Monogr 163: 251–276.
Setlow B, McGaugh JL (1998). Sulpiride infused into the nucleus accumbens posttraining impairs memory of spatial water maze training. Behav Neurosci 112: 603–610.
Setlow B, McGaugh JL (2000). D2 dopamine receptor blockade immediately post-training enhances retention in hidden and visible platform versions of the water maze. Learn Memory 7: 187–191.
Sheng P, Cerruti C, Cadet JL (1994). Methamphetamine (METH) causes reactive gliosis in vitro: attenuation by the ADP-ribosylation (ADPR) inhibitor, benzamide. Life Sci 55: PL51–PL54.
Smith-Roe SL, Kelley AE (2000). Coincident activation of NMDA and dopamine D1 receptors within the nucleus accumbens core is required for appetitive instrumental learning. J Neurosci 20: 7737–7742.
Sora I, Wichems C, takahashi N, Li XF, Zeng Z, Revay R et al (1998). Cocaine reward models: conditioned place preference can be established in dopamine- and in serotonin-transporter knockout mice. Proc Natl Acad Sci USA 95: 7699–7704.
Stephans SE, Yamamoto BK (1994). Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse 17: 203–209.
Varga V, Jenei Z, Janaky R, Saransaari P, Oja SS (1997). Glutathione is an endogenous ligand of rat brain N-methyl-D-aspartate (NMDA) and 2-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate (AMPA) receptors. Neurochem Res 22: 1165–1171.
Villemagne V, Yuan J, Wong DF, Dannals RF, Hatzidimitriou G, Mathews WB et al (1998). Brain dopamine neurotoxicity in baboons treated with doses of methamphetamine comparable to those recreationally abused by humans: evidence from [11C]WIN-35,428 position emission tomography studies and direct in vitro determinations. J Neurosci 18: 419–427.
Volkow ND, Chang L, Wang GJ, Fowler JS, Franceschi D, Sedler M et al (2001a). Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J Neurosci 21: 9414–9418.
Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D et al (2001b). Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry 158: 377–382.
Warr O, Takahashi M, Attwell D (1999). Modulation of extracellular glutamate concentration in rat brain slices by cystine-glutamate exchange. J Physiol 514: 783–793.
Weber GF (1999). Final common pathways in neurodegenerative diseases: regulatory role of the glutathione cycle. Neurosci Biobehav Rev 23: 1079–1086.
White NM, Carr GD (1985). The conditioned place preference is affected by two independent reinforcement processes. Pharmacol Biochem Behav 23: 37–42.
White NM, Packard MG, Hiroi N (1991). Place conditioning with dopamine D1 and D2 agonists injected peripherally or into nucleus accumbens. Psychopharmacol 103: 271–276.
Acknowledgements
This work was supported by RO1DA012867 and R01 DA019107 from the National Institute on Drug Abuse, National Institutes of Health. Cindy Achat-Mendes was a Lois Pope LIFE fellow. The authors thank Erica Givens for excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Achat-Mendes, C., Anderson, K. & Itzhak, Y. Impairment in Consolidation of Learned Place Preference Following Dopaminergic Neurotoxicity in Mice is Ameliorated by N-acetylcysteine but not D1 and D2 Dopamine Receptor Agonists. Neuropsychopharmacol 32, 531–541 (2007). https://doi.org/10.1038/sj.npp.1301119
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301119
Keywords
This article is cited by
-
Protective action of Grewia asiatica (phalsa) berries against scopolamine-induced deficit in learning and memory using behavior paradigms in rats
Advances in Traditional Medicine (2020)
-
Flood-conditioned place aversion as a novel non-pharmacological aversive learning procedure in mice
Scientific Reports (2018)
-
The toxic influence of dibromoacetic acid on the hippocampus and pre-frontal cortex of rat: involvement of neuroinflammation response and oxidative stress
Metabolic Brain Disease (2017)
-
Effect of an Inhibitor of Sphingomyelinases, N-Acetylcysteine, on Cognitive Functions in Old Rats
Neurophysiology (2014)
-
N-acetylcysteine protects against cadmium-induced germ cell apoptosis by inhibiting endoplasmic reticulum stress in testes
Asian Journal of Andrology (2013)
