
Abstract
Intense or chronic stress can produce pathophysiological alterations in the systems involved in the stress response. The amygdala is a key component of the brain's neuronal network that processes and assigns emotional value to life's experiences, consolidates the memory of emotionally significant events, and organizes the behavioral response to these events. Clinical evidence indicates that certain stress-related affective disorders are associated with changes in the amygdala's excitability, implicating a possible dysfunction of the GABAergic system. An important modulator of the GABAergic synaptic transmission, and one that is also central to the stress response is norepinephrine (NE). In the present study, we examined the hypothesis that stress impairs the noradrenergic modulation of GABAergic transmission in the basolateral amygdala (BLA). In control rats, NE (10 μM) facilitated spontaneous, evoked, and miniature IPSCs in the presence of β and α2 adrenoceptor antagonists. The effects of NE were not blocked by α1D and α1B adrenoceptor antagonists, and were mimicked by the α1A agonist, A61603 (1 μM). In restrain/tail-shock stressed rats, NE or A61603 had no significant effects on GABAergic transmission. Thus, in the BLA, NE acting via presynaptic α1A adrenoceptors facilitates GABAergic inhibition, and this effect is severely impaired by stress. This is the first direct evidence of stress-induced impairment in the modulation of GABAergic synaptic transmission. The present findings provide an insight into possible mechanisms underlying the antiepileptogenic effects of NE in temporal lobe epilepsy, the hyperexcitability and hyper-responsiveness of the amygdala in certain stress-related affective disorders, and the stress-induced exacerbation of seizure activity in epileptic patients.
Similar content being viewed by others

INTRODUCTION
Many components of the biological response to emotional stressors are of vital importance in enabling the individual to cope with stress. However, it is well known that excessive or repeated stress can have detrimental effects on health that are often associated with functional alterations in the systems involved in the stress response (Vermetten and Bremner, 2002; Vanitallie, 2002; McEwen, 2002; Pawlak et al, 2003). The amygdala is a key component of the brain's neuronal network that determines the emotional significance of external events (LeDoux, 1992; Davis, 1994; Breiter et al, 1996; Schneider et al, 1997; LaBar et al, 1998; Buchel et al, 1998; Whalen et al, 1998; Baird et al, 1999; Davidson et al, 1999; Davidson and Slagter, 2000; Buchel and Dolan, 2000). Via efferent pathways to the hypothalamus, the amygdala can also trigger the neuroendocrine cascades that are part of the stress response (Habib et al, 2001; Pitkänen, 2000; Davis, 1992) and via reciprocal connections with the cerebral cortex and limbic structures, it modulates the orchestration of the behavioral response (Goldstein et al, 1996; Pitkanen et al, 2000). Therefore, understanding the changes in the amygdala's physiology and function induced by stress is critical in understanding the pathophysiology of stress, and may aid the development of new therapeutic strategies for the prevention and treatment of stress-related, affective disorders.
Different lines of evidence point to the possibility that the function of the GABAergic system may be impaired by stress. First, in a number of brain regions, benzodiazepine receptor binding is altered by stress (Lippa et al, 1978; Medina et al, 1983; Miller et al, 1987; Weizman et al, 1989; Bremner et al, 2000). Second, in certain stress-related psychiatric disorders, the amygdala exhibits higher than normal levels of basal activity (Abercrombie et al, 1998; Drevets, 1999), or exaggerated responses to fearful stimuli (Rauch et al, 2000; Villarreal and King, 2001). Since the GABAergic system is a primary regulator of neuronal excitability, pathophysiological changes in GABAergic transmission may underlie the amygdala's hyper-responsiveness and hyperexcitability in these emotional disorders. Third, many psychotropic drugs that are effective in the treatment of emotional disorders target or influence GABAergic transmission. Fourth, stress exacerbates the frequency of seizures in epileptic patients (Temkin and Davis, 1984; Frucht et al, 2000). However, there is no direct evidence, so far, for stress-induced impairment in GABAergic synaptic transmission.
One of the modulators of GABA release is norepinephrine (NE), which is also central to the stress response. During stress, there is a dramatic increase in noradrenergic activity following the peripheral release of epinephrine from the adrenal glands, and the central release of NE, predominantly from the locus ceruleus (Stanford, 1995; Bremner et al, 1996). The amygdala receives dense noradrenergic afferents from the locus ceruleus (Pitkänen, 2000), as well as from other brain regions such as the nucleus of the solitary tract (Pitkänen, 2000; Clayton and Williams, 2000; Williams et al, 2000). During stress, there is a strong enhancement of NE release in the amygdala (Galvez et al, 1996; Stanford, 1995; Quirarte et al, 1998; Tanaka et al, 2000). The short- and long-term consequences of stress-induced excessive NE release on amygdala's physiology are unknown.
NE modulates GABAergic inhibition primarily via the α1 subtype of adrenergic receptors (Gellman and Aghajanian, 1993; Alreja and Liu, 1996; Bergles et al, 1996; Kawaguchi and Shindou, 1998). There is evidence suggesting that α1 adrenoceptors are affected by stress. Thus, chronic stress, in rats, reduces the expression of these receptors in the hypothalamus and brain stem (Miyahara et al, 1999). α1 adrenoceptor binding is also reduced in depressed patients (Crow et al, 1984; Gross-Isseroff et al, 1990), and blockade of these receptors in rats increases depressive behavior (Stone and Quartermain, 1999). The physiological implications of stress-induced reduction in α1 adrenoceptor activity are not known.
In the present study, we investigated whether NE modulates GABAergic transmission in the basolateral nucleus of the amygdala (BLA), and if so, whether the noradrenergic modulation of the GABAergic transmission is altered by exposure to stress. We studied the BLA because this amygdala region is heavily involved in the processing of emotional experiences, as it receives both direct and indirect thalamic and cortical inputs and is extensively interconnected with the prefrontal/frontal cortex and the hippocampus (Pitkänen, 2000). Furthermore, it appears that the BLA selectively (among the different amygdala nuclei) modulates the consolidation of emotional memories (Cahill and McGaugh, 1998; Ferry et al, 1999). Our results show that NE facilitates spontaneous, evoked, and action potential-independent, quantal GABA release in the BLA via the α1A subtype of adrenergic receptors, and that exposure to stress severely impairs this α1 adrenoceptor-mediated facilitation of GABA release.
METHODS
Animals and Stress Protocol
All animal experiments were performed in accordance with our institutional guidelines after obtaining the approval of the Institutional Animal Care and Use Committee (IACUC). Male, Sprague–Dawley rat pups were received with their mother at postnatal day (PND) 17, and housed in a climate-controlled environment on a 12 h light/dark cycle (lights on at 0700). On PND 21, the rats were weaned, assigned numbers, and randomly divided into control and stressed groups. They were housed individually, with food and water supplied ad libitum. The ‘stressed group’ was exposed to stress on PND 22, 23, and 24. The rats were killed and brain slices were prepared on PND 24 and 25. The experiments were performed in a blind manner. The investigators did not know whether they used a control or a stressed rat until the data were analyzed.
Stress exposure consisted of a 2-h per day session of immobilization and tail-shocks, for 3 consecutives days. The animals were stressed in the morning (between 0800 and 1200). They were restrained in a plexiglas tube, and 40 electric shocks (2 mA, 3 s duration) were applied at varying intervals (140–180 s). This stress protocol was adapted from the ‘learned helplessness’ paradigm in which animals undergo an aversive experience under conditions in which they cannot perform any adaptive response (Seligman and Maier, 1967; Seligman and Beagley, 1975). We stressed the rats for 3 consecutive days because it has been previously demonstrated that repeated stress sessions for 3 days is more effective than a single stress session in producing physiological and behavioral abnormalities, such as elevations in the basal plasma corticosterone levels, exaggerated acoustic startle responses, and reduced body weight (Servatius et al, 1995; Ottenweller et al, 1989). More stress sessions, beyond the 3 days, do not appear to produce greater physiological and behavioral changes (Servatius et al, 1995; Ottenweller et al, 1989).
Slice Preparation
Experimental procedures
The amygdala slice preparation has been described previously (Li et al, 2001). Briefly, the rats were anesthetized with halothane and then decapitated. The brain was rapidly removed and placed in an ice-cold artificial cerebrospinal fluid (ACSF) composed of (in mM) 125 NaCl, 2.5 KCl, 2.0 CaCl2, 1.0 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, and 11 glucose, bubbled with 95% O2/5% CO2. A block containing the amygdala region was prepared by rostral and caudal coronal cuts, and coronal slices, 400 μm thick, were cut using a Vibratome (series 1000, Technical Products International, St Louis, Missouri). Slices were kept in a holding chamber containing oxygenated ACSF at room temperature, and experiments started ⩾1 h after slice preparation.
Electrophysiology
For whole-cell recordings, slices were transferred to a submersion-type recording chamber where they were continuously perfused with oxygenated ACSF at a rate of 4 ml/min. All experiments were carried out at 32°C. Tight-seal (>1 GΩ) whole-cell recordings were obtained from the cell body of neurons in the BLA region. Patch electrodes were fabricated from borosilicate glass and had a resistance of 1.5–5.0 MΩ when filled with a solution containing (in mM) Cs-gluconate, 135; MgCl2, 10; CaCl2, 0.1; EGTA, 1; HEPES, 10; QX-314, 20; NaATP, 2; Na3GTP, 0.2 and Lucifer yellow, 0.4% (pH 7.3, 285–290 mOsm). Neurons were visualized with an upright microscope (Nikon Eclipse E600fn) using the Nomarski-type differential interference optics through a × 60 water immersion objective. Neurons with a pyramidal appearance were selected for recordings. During whole-cell recordings, neurons were filled passively with 0.4% Lucifer yellow (Molecular Probes, Eugene, Oregon) for post hoc morphological identification, as described previously (Braga et al, 2003). The fluorescence image of the dye-filled neurons was captured by a Leica DM RXA fluorescence microscope equipped with an SPOT2 digital camera and a laser scanning confocal microscope (Bio RAD, MRC-600). Neurons were voltage clamped using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Inhibitory postsynaptic currents (IPSCs) were pharmacologically isolated and recorded at a holding potential of −70 mV. Synaptic responses were evoked with sharpened tungsten bipolar stimulating electrodes (2 μm diameter, World Precision Instruments, Sarasota, Florida) placed in the BLA, 50–100 μm from the recording electrode. Stimulation was applied, at 0.1 Hz, using a photoelectric stimulus isolation unit having a constant current output (PSIU6, Grass Instrument Co., W. Warwick, RI). Access resistance (8–26 MΩ) was regularly monitored during recordings, and cells were rejected if it changed by more than 15% during the experiment. The signals were filtered at 2 kHz, digitized (Digidata 1322A, Axon Instruments, Inc.), and stored on a computer using the pCLAMP8 software (Axon Instruments, Inc.). The peak amplitude, 10–90% rise time, and the decay time constant of IPSCs were analyzed off-line using pCLAMP8 software (Axon Instruments) and the Mini Analysis Program (Synaptosoft, Inc., Leonia, NJ). Miniature IPSCs (mIPSCs) were analyzed off-line using the Mini Analysis Program (Synaptosoft, Inc., Leonia, NJ), and detected by manually setting the threshold for each mIPSC after visual inspection.
For field potential recordings, slices were transferred to an interface-type recording chamber maintained at 32°C, where they were perfused with ACSF at 0.7–1 ml/min. Field potentials were recorded in the BLA, while stimulation was applied to the external capsule, at 0.05 Hz (Aroniadou-Anderjaska et al, 2001). Recording glass pipettes were filled with 2 N NaCl (2–5 MΩ). Bipolar stimulating electrodes were constructed from twisted, stainless-steel wires, 50 μm in diameter. The field potentials were filtered at 1 kHz, and digitized on-line at 5 kHz.
All data are presented as mean±SEM. For body weight data, sample size n refers to the number of rats. For electrophysiological experiments, sample size n refers to the number of slices. This corresponds to the number of neurons, in whole-cell recordings, as a single neuron was studied from each slice. From each rat, two slices were used for each type of experiment (whole-cell recordings or field potential recordings). The results were tested for statistical significance using the Student's paired t-test.
Drugs
The following drugs were used: D-(−)-2-amino-5-phosphonopentanoic acid (D-AP5, Tocris Cookson, Ballwin, Missouri), an NMDA receptor antagonist; 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, Tocris Cookson, Ballwin, Missouri), a potent AMPA/kainate receptor antagonist; (2S)-(+)-5,5-dimethyl-2-morpholineacetic acid (SCH50911, Tocris Cookson, Ballwin, Missouri), a GABAB receptor antagonist; bicuculline methiodide (Sigma), a GABAA receptor antagonist; tetrodotoxin (TTX, Sigma), a sodium channel blocker; DL-propranolol (Sigma), a β adrenoceptor antagonist; (1-[4-amono-6,7-dimethoxy-2-quinazolinyl]-4-[2-furanylcarbonyl]-piperazine hydrochloride (prazosin, Sigma), an α1 adrenoceptor antagonist; yohimbine hydrochloride (Sigma), an α2 adrenoceptor antagonist; N-[5-(4,5-dihydro-1H-imidazol-2-yl)-2-hydroxy-5,6,7,8-tetrahydro-naphthalen-1-yl]methanesulfonamide hydrobromide (A61603, Tocris Cookson, Ballwin, Missouri), a selective α1A agonist (Knepper et al, 1995); chloroethylclonidine (CEC, Sigma), an irreversible antagonist that blocks both α1B and α1D adrenoceptors (Xiao and Jeffries, 1998); 8-[2-[4-(2-methoxyphenyl)-1-piperazinil]ethyl]-8-azaspiro[4,5]decane-7,9-dione dihydrochloride (BMY 7378, Tocris Cookson, Ballwin, Missouri), a selective antagonist of α1D adrenoceptors (Deng et al, 1996; Saussy Jr et al, 1996); 2-(2,6-dimethoxyphenoxyethy)aminomethyl-1,4-benzodioxane hydrochloride (WB4101, Tocris Cookson, Ballwin, Missouri), a selective antagonist of the α1A adrenoceptor (Zhong and Minneman, 1999).
RESULTS
The body weight of the control and stressed rats was measured daily between 1400 and 1500. The control rats were 44.5±1.5 g (n=24) on PND 21 and 58.8±1.9 g (n=24) on PND 24 (Figure 1). The body weight of the stressed group was 44.2±1.8 g (n=23) before the first stress session on PND 21, and 51.0±2.3 g (n=20) after the last stress session, on PND 24. The difference in body weight between stressed and control rats was statistically significant after the second day of stress (p<0.01). Stressed rats that were not used for electrophysiological experiments continued to display reduced body weight gain for as long as body weight was monitored (up to 10 days after stressor cessation, data not shown).

Restrain/tail-shock stress reduces body weight gain. Exposure to stress on PNDs 22, 23, and 24 reduced body weight gain. The body weight difference between control and stressed rats was statistically significant after the first day of stress (**p<0.01). Data on PND 26 are from rats that were not used for electrophysiological experiments. Sample sizes range from 12 (PND 26) to 24 rats.
Stress Blocks Noradrenergic Facilitation of GABAergic Synaptic Transmission
Noradrenergic modulation of spontaneous IPSCs (sIPSCs)
To investigate whether NE modulates GABAergic transmission in the BLA, and whether stress alters this modulation, we first examined the effects of NE on action-potential dependent, sIPSCs recorded from BLA pyramidal neurons, in control and stressed rats. sIPSCs were recorded at a holding potential of −70 mV, and in the presence of D-AP5 (50 μM), CNQX (10 μM), propranolol (10 μM), and yohimbine (20 μM) to block NMDA, AMPA/kainate, and β and α2 receptors, respectively. In control rats, the mean frequency of sIPSCs recorded from the soma of BLA pyramidal neurons was 3.1±1.6 Hz (n=21). Bath application of bicuculline (10 μM) eliminated sIPSCs, confirming that they were mediated by GABAA receptors. NE at 1, 10, and 100 μM produced a dose-dependent enhancement in the frequency and amplitude of sIPSCs (Figure 2). At 100 μM of NE, the enhancement of sIPSCs was too high to be quantified precisely. The 10 μM concentration appeared to be close to the EC50, and therefore it was used in subsequent experiments. After the application of 10 μM NE, the mean frequency of sIPSCs was increased to 984.39±148.2% of the control values (n=21, p<0.01; Figure 3a). The amplitude of sIPSCs was increased to 144.0±12.8% of the control values (n=21, p<0.05; Figure 3a). These effects persisted throughout the application of NE and were completely reversed after removal of the agonist. The effects of NE were not accompanied by any significant change in the rise time or decay time constant of sIPSCs (Figure 3a), and were blocked by the α1 adrenoceptor antagonist prazosin (1 μM, Figure 3c), confirming that NE was acting via α1 adrenergic receptors.

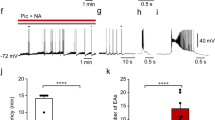
Activation of α1 adrenoceptors increases tonic inhibition of BLA pyramidal neurons in a dose-dependent manner. (a–c) sIPSCs recorded from three different cells are shown. The holding potential is −70 mV. The medium contains D-AP5 (50 μM), CNQX (10 μM), propranolol (10 μM), and yohimbine (20 μM). The application of 1, 10, and 100 μM NE increased the frequency of sIPSCs in a dose-dependent manner. The bar graph (d) shows group data of the increase of sIPSC frequency (n=8 for each concentration of NE, **p<0.01). (e) Photomicrograph of pyramidal cell (b) showing the typical morphology of the recorded neurons. The cell has been labeled with Lucifer Yellow. Scale bar, 40 μm.

Activation of α1 adrenoceptors increases tonic inhibition of BLA pyramidal neurons in control rats, but not in stressed rats. (a) Top trace: effects of NE (10 μM) on sIPSCs recorded from a BLA pyramidal cell of a control rat. The holding potential is −70 mV. The medium contains D-AP5 (50 μM), CNQX (10 μM), propranolol (10 μM), and yohimbine (20 μM). Middle graphs: cumulative probability plots of interevent intervals and amplitude of sIPSCs, in control conditions and during NE perfusion (same cell as in the top trace). Bottom graphs: pooled data (mean±SEM) from 21 neurons. The bar graph on the left shows the NE-induced changes in amplitude, frequency, and kinetics of sIPSCs. The bar graph on the right panel shows the time course of changes in sIPSC frequency during the application of NE. *p<0.05, **p<0.01. (b) Top trace: sIPSCs recorded from a BLA pyramidal cell of a stressed rat (the holding potential is −70 mV); NE (10 μM) had no significant effect. Middle graphs: cumulative probability plots of interevent intervals and amplitude of sIPSCs in control conditions and during NE perfusion (same cell as in the top trace). Bottom graphs: pooled data (mean±SEM) from 19 neurons. Effects of NE on the amplitude, kinetics, and frequency of sIPSCs in stressed rats. (c) Prazosin (1 μM) prevented the NE-induced increase of sIPSCs observed in control rats. (d) The bar graph shows the effects of NE on the mean frequency of sIPSCs recorded from control rats (in the absence and in the presence of prazosin), and stressed rats (in the absence of prazosin). *p<0.05, **p<0.01.
In stressed rats, the mean frequency of sIPSCs was 2.6±2.3 Hz. NE (10 μM) had no significant effect on the frequency or amplitude of sIPSCs. Thus, in the presence of NE (10 μM), the frequency of sIPSCs was 128.9±19.2% and the amplitude was 111.4±10.2% of the control values (n=19, Figure 3b). In addition, bath perfusion of NE (10 μM) caused no significant changes in the kinetics of these currents (rise time and decay time constant of sIPSCs; Figure 3b).
To identify the subtype of α1 adrenoceptors involved in the effects of NE on control rats, we first applied NE (10 μM) in the additional presence of CEC (10 μM) and BMY 7378 (300 nM) to block α1B and α1D adrenoceptors. There was no significant attenuation of the effects of NE in the presence of these antagonists (Figure 4). Thus, NE increased the frequency of sIPSCs from 2.8±2.4 to 27.1±7.9 Hz (p<0.01, n=6; Figure 4), and the amplitude of sIPSCs to 154±11.3% of the control values (p<0.05, n=6; Figure 4).

The NE-induced enhancement of sIPSCs is not blocked by α1B and α1D adrenoceptor antagonists. Top trace: sIPSCs recorded from a BLA pyramidal cell of a control rat (holding potential is −70 mV). Bath application of NE (10 μM) in the presence of D-AP5 (50 μM), CNQX (10 μM), propranolol (10 μM), yohimbine (20 μM), CEC (10 μM), and BMY 7378 (300 nM) reversibly increased the frequency and amplitude of sIPSCs. The bar graph shows pooled data (mean±SEM) from six neurons. *p<0.05, **p<0.01.
Next, we examined the effects of the specific α1A adrenoceptor agonist A61603. In control rats, A61603 (1 μM) increased the frequency and amplitude of sIPSC to 1034±158.6 and 162±14.2% of the control values, respectively (p<0.01, n=16; Figure 5a). There were no effects on the rise time or the decay time constant of sIPSCs (Figure 5a). In stressed rats, A61603 had no significant effect (Figure 5b). Thus, in the presence of 1 μM A61603 the frequency of sIPSCs was 132±21% and the amplitude of sIPSCs was 106±8.8% of the control values (n=18, Figure 5b). The effects of A61603 on sIPSCs in control rats were blocked by the selective α1A adrenoceptor antagonist WB4101 (1 μM, Figures 5c and d).

Activation of α1A adrenoceptors increases tonic inhibition of BLA pyramidal neurons in control rats, but not in stressed rats. (a) Top trace: sIPSCs recorded from a BLA pyramidal cell of a control rat (the holding potential is −70 mV). Bath application of A61603 (1 μM), a specific α1A adrenoreceptor agonist, reversibly increased the frequency and amplitude of sIPSCs. The slice medium contains D-AP5 (50 μM), CNQX (10 μM), propranolol (10 μM), and yohimbine (20 μM). Middle graphs: cumulative probability plots of sIPSC interevent intervals and amplitude in control conditions and during A61603 perfusion (same cell as in the top trace). Bottom graphs: bar graphs show pooled data (mean±SEM) from 16 neurons. (b) sIPSCs recorded from a BLA pyramidal cell of a stressed rat (the holding potential is −70 mV). Bath application of A61603 (1 μM) caused no significant change in the frequency or amplitude of sIPSCs. Middle graphs: cumulative probability plots of sIPSCs interevent intervals and amplitude in control conditions and during A61603 (1 μM) perfusion (same cell as in the top trace). Bottom graphs: bar graph shows pooled data (mean±SEM) from 18 neurons. (c) WB4101 (1 μM) prevented the A61603-induced effects observed in control rats. (d) Bar graph shows the effects of A61603 (1 μM) on the mean frequency of sIPSCs recorded from control rats (in the absence and in the presence of WB4101), and stressed rats (in the absence of WB4101). *p<0.05, **p<0.01.
Taken together, these results suggest that (1) NE, acting via α1A adrenoceptors, enhances tonic inhibition of pyramidal cells in the BLA by inducing a massive increase in action potential-dependent spontaneous release of GABA, and (2) stress impairs this function of NE.
Noradrenergic modulation of evoked IPSCs (eIPSCs)
It has been shown previously that NE reduces evoked inhibitory transmission in the hippocampus via α adrenoceptors (Madison and Nicoll, 1988; Doze et al, 1991). More recently, in the sensorimotor cortex, it was found that NE actually has a small facilitatory effect on eIPSCs, which is detected when GABAB receptors are blocked (Bennett et al, 1997). To determine the effects of NE on evoked inhibitory transmission in the BLA we applied 10 μM NE while recording eIPSCs in control rats. In the absence of a GABAB receptor antagonist, NE (10 μM) reduced the amplitude of eIPSCs to 48.2±10.3% of the control levels (p<0.01, n=8; Figure 6). However, in the presence of SCH50911 (20 μM), a specific antagonist of the GABAB receptors, NE enhanced the amplitude of eIPSCs to 162.4±9.3% of the control, p<0.01, n=10; Figure 7a) without affecting the rise time and decay time constant of the eIPSCs (Figure 7a). Similar effects were obtained when α1A adrenoceptors were activated by the application of 1 μM A61603 (Figure 7c). Thus, A61603 (1 μM) increased the amplitude of eIPSCs to 159.4±10.7% of the control (p<0.01, n=8, Figure 7c) without affecting the kinetics of the eIPSCs (Figure 7c). The effects of the drugs were reversible. In stressed rats, neither NE nor A61603 had a significant effect on the amplitude, rise time, and decay time constant of eIPSCs (Figure 7b and d). In the presence of NE (10 μM), the eIPSC amplitude was 109±8.2% of the control (n=11), and in the presence of A61603, the amplitude of the eIPSCs was 103±7.4 % of the control (n=10). These results suggest that (1) NE facilitates evoked the GABAergic transmission via α1A adrenergic receptors, (2) this facilitatory effect is masked due to the activation of presynaptic GABAB autoreceptors following the NE-induced enhancement of spontaneous GABA release, and (3) stress blocks the facilitatory effect of NE on evoked GABA release.

Activation of α1 adrenoceptors reduces the amplitude of eIPSCs in control rats. Top traces: eIPSCs recorded from a BLA neuron of a control rat. The slice medium contains D-AP5 (50 μM), CNQX (10 μM), propranolol (10 μM), and yohimbine (20 μM). NE reduced the amplitude of eIPSCs with no significant effect on their kinetics. Bottom graphs: the plot shows the time course of the NE effects on the amplitude of eIPSCs (same cell as in top traces). The bar graph shows the relative (% of control) NE-induced changes in amplitude and kinetics of eIPSCs. Pooled data from eight neurons. **p<0.01.

In the presence of a GABAB receptor antagonist, activation of α1A adrenoceptors increases the amplitude of eIPSCs in control rats, but not in stressed rats. (a) Top traces: eIPSCs recorded from a BLA pyramidal cell of a control rat. In addition to D-AP5 (50 μM), CNQX (10 μM), propranolol (10 μM), and yohimbine (20 μM), the slice medium also contains 20 μM SCH50911. NE increased the amplitude of the eIPSCs, without affecting their kinetics. Bottom graphs: the plot shows the time course of the NE effect on eIPSC amplitude (same cell as in the top traces). The bar graph shows the effect of NE on the amplitude and kinetics of eIPSCs. Pooled data from 10 neurons. **p<0.01. (b) Data similar to those shown in (a), but from stressed rats. The bar graph shows pooled data from 11 neurons. (c) In control rats, the α1A agonist A61603 produced similar effects to those of NE. Top traces and bottom left plot show data from the same cell. The bar graph shows pooled data from eight BLA neurons. (d) In stressed rats, A61603 had no significant effects on eIPSCs. The bar graph shows pooled data from 10 BLA neurons.
Noradrenergic modulation of mIPSCs
The enhancement of eIPSCs and action-potential-dependent sIPSCs by NE could be due to a depolarizing effect via the activation of somatodendritic α1A adrenoceptors on GABAergic neurons, and/or due to a direct effect at GABAergic terminals. To determine whether NE modulates GABA release by a direct effect on GABAergic terminals in the BLA, we tested the effects of NE on mIPSCs, which do not depend on the presynaptic invasion of action potentials or Ca2+ influx. mIPSCs were recorded in a medium containing D-AP5 (50 μM), CNQX (10 μM), propranolol (10 μM), yohimbine (20 μM), and TTX (1 μM). In the absence of NE, the frequency of mIPSCs was 0.68±0.32 Hz and their amplitude was 114.0±12 pA (n=10). NE (10 μM) increased the frequency of mIPSCs to 182.3±9.6 % of the control levels (p<0.01, n=10; Figure 8a). The amplitude, rise time, and decay time constant of the mIPSCs were not significantly affected by 10 μM NE (Figure 8a). Similar effects were observed after the application of the α1A-specific agonist A61603 (Figure 8c). A61603 (1 μM) increased the frequency of mIPSCs from 0.71±0.24 to 1.28±0.31 (178±12.4% of the control, p<0.01, n=9; Figure 8c). The amplitude and kinetics of mIPSCs were not affected by A61603 (Figure 8c).

Activation of α1A adrenoceptors increases the frequency of mIPSCs in control rats, but not in stressed rats. mIPSCs were recorded in the presence of TTX (1 μM), D-AP5 (50 μM), CNQX (10 μM), propranolol (10 μM), and yohimbine (20 μM). (a) Top traces: mIPSCs recorded from a BLA pyramidal neuron of a control rat. NE (10 μM) increased the frequency of mIPSCs. Bottom graph: the left panel shows the cumulative probability plots of interevent intervals of mIPSCs under control conditions and during the application of NE (same cell as in top traces). The bar graph shows the effect of NE on the amplitude, kinetics, and frequency of mIPSCs. Pooled data from 10 neurons, **p<0.01. (b) Similar data to those shown in (a), but from stressed rats. NE had no significant effect on mIPSCs. The bar graph shows pooled data from 10 neurons. (c) In control rats, the α1A antagonist A61603 had similar effects to those induced by NE. The bar graph shows pooled data from nine BLA neurons. (d) A61603 had no significant effects on mIPSCs recorded from BLA pyramidal cells of stressed rats. The bar graph shows pooled data from eight cells.
In stressed rats, neither NE (10 μM) nor A61603 (1 μM) produced a significant effect on mIPSCs frequency, amplitude, or kinetics (Figure 8b and d). Thus, the frequency of mIPSCs was 0.68±0.25 and 0.64±0.34 Hz before and during the application of NE, respectively (n=10), and 0.72±0.27 and 0.63±0.31 Hz in the presence and absence of 1 μM A61603, respectively (n=8).
These results suggest that (1) NE facilitates GABA release by a direct effect on GABAergic terminals, and (2) this mechanism of noradrenergic facilitation of GABA release is impaired by stress.
Facilitation of GABAergic Transmission by α1A Adrenoceptors is Mediated by Phospholipase C (PLC)
Studies in other brain regions or cell types have shown that α1 adrenoceptors are coupled to PLC via a G-protein, and can increase the intracellular calcium concentration [Ca2+]i by mobilizing Ca2+ from intracellular stores, as well as by increasing the Ca2+ influx (Schwinn et al, 1991; Wu et al, 1992; Cohen and Almazan, 1993; Lepretre et al, 1994; Kulik et al, 1999). However, certain effects of α1A activation involve signaling pathways that are independent of PLC activation and intracellular Ca2+ rise (Berts et al, 1999). To determine whether the α1A adrenoceptor-mediated facilitation of GABA release, in the BLA, involves the activation of PLC, we examined whether the effects of NE on the GABAergic transmission are blocked by a PLC inhibitor. In control rats, NE (10 μM) or A61603 (1 μM) enhanced the frequency and amplitude of sIPSCs in the presence of U73343 (20 μM), the inactive isomer of the PLC inhibitor U73122, but had no effects in the presence of 20 μM U73122 (Figure 9). Thus, in the presence of U73343, NE increased the frequency of sIPSCs to 1022.8±105.3% of the control levels (p<0.01, n=8; Figure 9a) and increased the amplitude of sIPSCs to 161±11.7% (p<0.01, n=6; Figure 9a); A61603 (1 μM) increased the frequency of sIPSCs to 978.1±102.1% (p<0.01, n=8; Figure 9b), and increased the amplitude of sIPSCs to 154±12.3% of the control levels (p<0.01, n=8; Figure 9b). In contrast, in the presence of U73122 (20 μM), NE (10 μM) and A61603 (1 μM) failed to induce any significant changes in the frequency and amplitude of sIPSCs (Figure 9c–e). Similarly, the effects of NE (10 μM) on the amplitude of eIPSCs, as well as on the frequency of mIPSCs, were blocked by 20 μM U73122 (not shown).

α1A adrenoceptors in the BLA are coupled to PLC. (a–d) sIPSCs recorded from BLA pyramidal neurons. NE (a) or A61603 (b) increased the frequency and amplitude of sIPSCs in the presence of the inactive isomer of a PLC inhibitor (U73343), but had no effect in the presence of the PLC inhibitor U73122 (c and d). The slice medium contains D-AP5 (50 μM), CNQX (10 μM), propranolol (10 μM), and yohimbine (20 μM). (e) Bar graphs showing the effects of NE (10 μM) or A61603 (1 μM) on the frequency of sIPSCs in the presence of U73343 or U73122. Pooled data from eight neurons.
Stress Blocks α1A Adrenoceptor-Mediated Suppression of BLA Field Potentials
Since the activation of α1A adrenoceptors facilitates GABAergic transmission, the function of these receptors at the network level could be to dampen neuronal excitability and responsiveness. However, while spontaneous GABAergic activity is dramatically enhanced by activation of α1A adrenoceptors (Figure 5), evoked GABAergic transmission is suppressed due to presynaptic inhibition of GABA release via GABAB autoreceptors (Figure 6). Therefore, under physiological conditions when GABAB receptors are not blocked, α1A adrenoceptor activation could enhance the amygdala's responsiveness (due to the reduction in evoked GABA release), unless the enhancement of spontaneously released extracellular GABA plays a more decisive role in neuronal excitability. To determine the net effect of α1A adrenoceptor activation on neuronal responsiveness and excitability in the BLA, and whether this effect is altered by stress, we investigated the effects of NE or A61603 on population, field responses, in the absence of GABAB receptor blockade, in control and stressed rats.
Field potentials in the BLA were evoked by stimulation of the external capsule. These responses consist of one major, negative component that corresponds in time course to the EPSP recorded intracellularly from BLA pyramidal cells (Aroniadou-Anderjaska et al, 2001; Chen et al, 2003), and is mediated by AMPA/kainate receptors (Aroniadou-Anderjaska et al, 2001). In control rats, 10 μM NE, in the presence of propranolol (10 μM) and yohimbine (20 μM), produced a significant reduction in the peak amplitude of evoked field potentials (83.8±5.3% of control levels, n=14, p<0.05; Figure 10a). Similarly, bath application of 1 μM A61603 caused a significant reduction in the peak amplitude of the field potentials to 83.1±5.2% of the control levels (p<0.05, n=12; Figure 10b). In contrast, in stressed rats, neither NE (10 μM) nor A61603 (1 μM) had a significant effect on the amplitude of the field potentials (Figure 10, bottom panels).

Activation of α1A adrenoceptors reduces BLA field potentials in control rats, but not in stressed rats. (a) Changes in the peak amplitude of BLA field potentials evoked by stimulation of the external capsule, in response to bath application of 10 M NE, in control (top panel, n=9) and stressed (bottom panel, n=10) rats. The medium contains propranolol (10 M) and yohimbine (20 M). (b) Similar data to those in (a), except that A61603 is applied in place of the NE. Pooled data from 10 slices (control rats, top panel) and eight slices (stressed rats, bottom panel). The slice medium same as in (a). Asterisks over error bars denote statistically significant reduction (p<0.05).
These results suggest that the function of α1A adrenoceptors in the BLA is to reduce neuronal excitability/responsiveness, and this function is impaired by stress.
DISCUSSION
The present study describes two main findings. First, activation of the α1A subtype of adrenergic receptors facilitates both tonic and phasic GABAA receptor-mediated inhibition of BLA pyramidal neurons. Second, stress produces a severe impairment of the α1A adrenoceptor-mediated facilitation of GABAergic synaptic transmission in the BLA. These findings provide one possible explanation for (1) the antiepileptic effects of NE in temporal lobe epilepsy, (2) the amygdala's hyperexcitability in stress-related affective disorders, and (3) the stress-induced increase in the frequency of seizures in epileptic patients.
NE Facilitates GABAergic Transmission in the BLA via presynaptic α1A Adrenoceptors
All three subtypes of α1 adrenoceptors, α1A, α1B, and α1D, are present in the amygdala, as determined by in situ hybridization (Day et al, 1997). The distribution of these receptors varies in different nuclei of the amygdala. The BLA expresses the α1A adrenoceptor subtype almost exclusively (Day et al, 1997; Domyancic and Morilak, 1997). The role of these receptors in the amygdala's physiology and function has been unknown. In the present study, we show that NE, acting via the α1A subtype of adrenergic receptors, facilitates GABA release in the BLA. Spontaneous, evoked, and quantal release of GABA were enhanced by NE or the specific α1A adrenoceptor agonist A61603.
Endogenous NE released from noradrenergic terminals reaches its targets both by diffusion and via conventional synapses (Papadopoulos and Parnavelas, 1990; Seguela et al, 1990; Asan, 1993; Arce et al, 1994; Li et al, 2002). In the BLA, noradrenergic axons form asymmetric synapses with the dendrites of GABAergic neurons (Li et al, 2002). Although α1A adrenoceptors may be located in such dendritic synapses and could be involved in the enhancement of spontaneous and evoked GABA release by NE, the increase in the frequency of mIPSCs by α1A adrenoceptor activation indicates the presence of these receptors on GABAergic terminals. The enhancement of spontaneous GABA release by NE has also been observed in other brain regions (Madison and Nicoll, 1988; Doze et al, 1991; Gellman and Aghajanian, 1993; Alreja and Liu, 1996; Bergles et al, 1996; Bennett et al, 1997, 1998; Kawaguchi and Shindou, 1998), and it is mediated via α1 adrenoceptors (Gellman and Aghajanian, 1993; Alreja and Liu, 1996; Bergles et al, 1996; Kawaguchi and Shindou, 1998); the specific α1 receptor subtype involved has not been determined. At least in the CA1 hippocampal area, it appears that α1 adrenoceptors are located only on somatodendritic regions of GABAergic cells, since mIPSCs are unaffected by adrenergic agonists (Bergles et al, 1996). Thus, the amygdala and the hippocampus may differ in the subcellular distribution of α1 adrenoceptors mediating the facilitation of GABA release.
Evoked GABA release in the hippocampus is suppressed by NE, and this effect is also mediated via α adrenoceptors (Madison and Nicoll, 1988). However, a similar effect of NE in the sensorimotor cortex has been found to be due to the activation of presynaptic GABAB autoreceptors; when GABAB receptors were blocked, NE enhanced evoked GABAergic transmission (Bennett et al, 1997). Similarly, in the present study, the facilitatory effect of NE on evoked GABAergic transmission was revealed only when GABAB receptors were blocked, suggesting that the accumulation of extracellular GABA due to the NE-induced enhancement of spontaneous GABA release inhibited evoked GABA release.
Since NE enhances spontaneous GABA release, but suppresses evoked GABA release when GABAB receptors are functional, this raises the question of what would be the net effect of α1A adrenoceptor activation on the overall excitability and responsiveness of the amygdala. The BLA field potentials were reduced by NE or A61603 in the absence of GABAB receptor antagonists. It is unlikely that this effect is due to a reduction in glutamate release, because glutamatergic transmission in the BLA is suppressed via α2, but not α1 adrenoceptor activation (Ferry et al, 1997). Thus, the reduction of the BLA field potentials by NE or A61603 suggests that the dramatic enhancement of spontaneously released GABA induced by α1A adrenoceptor activation (Figure 5) over-rides the reduction in evoked GABAergic transmission (Figure 7) producing a suppression of the amygdala's excitability.
The intracellular signaling mechanisms that mediate the physiological effects of α1A adrenoceptor activation in the BLA involve the activation of PLC, since a PLC inhibitor prevented the enhancement of sIPSCs, eIPSCs, and mIPSCs by NE and A61603. The activation of PLC may lead to mobilization of Ca2+ from intracellular stores, and/or Ca2+ influx, following phosphoinositide hydrolysis and formation of IP3, as it has been observed in different tissues and cell types following α1 adrenoceptor activation (Schoepp and Rutledge, 1985; Schwinn et al, 1991; Perez et al, 1993; Kulik et al, 1999; Zhong and Minneman, 1999; Khorchid et al, 2002), or α1A adrenoreceptor activation (Cohen and Almazan, 1993; Lepretre et al, 1994). In the present study, since NE or A61603 enhanced the frequency of mIPSCs, the influx of Ca2+ through voltage-gated calcium channels is not necessary for the α1A adrenoreceptor-mediated facilitation of GABA release in the BLA.
The amygdala is a key player in the pathogenesis and symptomatology of temporal lobe epilepsy (Gloor, 1992; Weiss et al, 2000; Avoli et al, 2002). NE has long been known to display anticonvulsant properties, but little is known about the underlying mechanisms (Chen et al, 1954; Stanton, 1992; Stanton et al, 1992; Szot et al, 1999; Stoop et al, 2000; Weinshenker et al, 2001) The α1A adrenoreceptor-mediated facilitation of GABA release in the BLA may be one of the mechanisms involved in the antiepileptic effects of NE in temporal lobe seizure disorders.
Stress Impairs the Function of α1A Adrenoceptors in the BLA
Previous studies have suggested that excessive or repeated stress can produce long-lasting alterations in the amygdala's structure and function. Thus, chronic immobilization, in rats, induces hypertrophy of the dendritic arborizations of pyramidal and stellate neurons in the BLA (Vyas et al, 2002; Pawlak et al, 2003). Fear conditioning or other types of stressors such as exposure to a predator produce long-lasting changes in the efficacy of synaptic transmission in the amygdala (LeDoux, 1992; Davis et al, 1994; Rogan et al, 1997, McKernan and Shinnick-Gallagher, 1997; Adamec et al, 2001). In human patients with stress-related affective disorders, the amygdala exhibits hypertrophy (Strakowski et al, 1999; Altshuler et al, 2000), increased levels of basal activity (Drevets, 1999), or exaggerated responses to fearful stimuli (Rauch et al, 2000). In the present study, repeated restrain/tail-shock stress produced a severe impairment in the α1A adrenoreceptor-mediated facilitation of GABA release in the BLA, indicating that stress impairs the function of α1A adrenoceptors. This impairment could result from receptor desensitization, internalization, or downregulation, or by an effect on the intracellular signaling pathways activated by PLC. In other brain regions, repeated stress reduces mRNA levels of α1 adrenoceptors (Miyahara et al, 1999). Adrenergic receptors desensitize or undergo downregulation following prolonged exposure to the agonist (Yang et al, 1999; Chalothorn et al, 2002). Thus, during stress exposure, excessive release of NE in the amygdala (Galvez et al, 1996; Quirarte et al, 1998; Tanaka et al, 2000) may be responsible for the impairment of the α1A adrenoreceptor function. In addition, previous studies have shown that restrain/tail-shock stress elevates plasma corticosterone levels (Servatius et al, 1995). Glucocorticoid receptors colocalize with α1 adrenoceptors (Fuxe et al, 1985; Williams et al, 1997), and it has been demonstrated that corticosterone downregulates α adrenoceptors (Stone et al, 1986, 1987; Joels and de Kloet, 1989). Therefore, another possibility is that the corticosterone released during exposure to stress downregulates α1A adrenoceptors. An important question is whether the impairment in the α1A adrenoceptor function is a transient or a long-term effect. The investigations described here focus on changes measured within a relatively short period of time after stressor cessation. However, preliminary experiments have revealed differences in the α1A adrenoceptor function between stressed and control rats on the fifth day after the termination of stress exposure, suggesting that the stress-induced dysfunction in the noradrenergic modulation of GABA release is not likely to be a short-term effect.
Functional implications
What are the possible functional implications of a stress-induced loss of the α1A adrenoceptor-mediated noradrenergic facilitation of GABA release in the BLA? In the normal amygdala, basal levels of NE, acting via α1A adrenoceptors, may contribute to tonic inhibition of BLA pyramidal neurons, by facilitating both action potential-dependent and -independent GABA release. The loss or impairment of this facilitation would result in hyperexcitability at rest, and a lower threshold of activation. When the normal amygdala is activated in response to an emotionally significant event triggering the release of NE, activation of α1A adrenoceptors will facilitate the role of inhibitory transmission in active neuronal circuits; this role is not only to prevent overexcitation, but also to shape and sharpen the flow of excitatory activity. Therefore, loss of the α1A adrenoceptor-mediated facilitation of synaptic inhibition may result in inappropriate overactivation of the amygdala and impairment in the processing and interpretation of an emotional stimulus. A dysfunction of this nature may also affect the formation of emotional memories. In the normal amygdala, noradrenergic facilitation of GABAergic transmission may either suppress memory formation (due to the suppression of excitation), or facilitate optimal registration of the memory trace (by regulating the level and flow of excitatory activity). In a hyper-responsive amygdala, when noradrenergic facilitation of GABA release is impaired, events of little emotional significance may be registered as significant, and memories of emotionally significant events may be ‘overconsolidated’. It should be noted, however, that the net effect of stress on the function of the noradrenergic system in the BLA remains to be determined, as stress may also induce changes in the interaction of NE with other adrenoceptor subtypes (β and α2) or neurotransmitter systems.
It has been hypothesized that the hyperactivity and hyper-responsiveness of the amygdala associated with certain affective disorders, such as PTSD, is due to the loss of proper cortical modulation of the amygdala, and/or due to an intrinsic lower threshold of amygdala response to emotionally significant stimuli (Villarreal and King, 2001). The present findings suggest that a reduction in GABAergic transmission due to the loss of the α1A adrenoceptor-mediated facilitation of GABA release may be one of the mechanisms responsible for the apparently reduced threshold of amygdala's activation in these affective disorders. The present findings also suggest that a stress-induced impairment in the function of α1A adrenoceptors, which could result in reduced tonic inhibition in the BLA, may be one of the mechanisms underlying the stress-induced increased frequency of seizures in patients with temporal lobe epilepsy (Temkin and Davis, 1984; Frucht et al, 2000). Moreover, our results suggest that the reduced central α1 adrenoceptor responsiveness (Asnis et al, 1985, 1992), and binding (Crow et al, 1984; Gross-Isseroff et al, 1990) in depressed patients may be stress-related, and that one of the physiological consequences of this reduction is an impaired modulation of the GABAergic transmission.
References
Abercrombie HC, Schaefer SM, Larson CL, Oakes TR, Lindgren KA, Holden JE et al (1998). Metabolic rate in the right amygdala predicts negative affect in depressed patients. Neuroreport 9: 3301–3307.
Adamec RE, Blundell J, Collins A (2001). Neural plasticity and stress induced changes in defense in the rat. Neurosci Biobehav Rev 25: 721–744.
Alreja M, Liu W (1996). Noradrenaline induces IPSCs in rat medial septal/diagonal band neurons: involvement of septohippocampal GABAergic neurons. J Physiol 494: 201–215.
Altshuler LL, Bartzokis G, Grieder T, Curran J, Jimenez T, Leight K et al (2000). An MRI study of temporal lobe structures in men with bipolar disorder or schizophrenia. Biol Psychiatry 48: 147–162.
Arce EA, Bennett-Clarke CA, Rhoades RW (1994). Ultrastructural organization of the noradrenergic innervation of the superficial gray layer of the hamster's superior colliculus. Synapse 18: 46–54.
Aroniadou-Anderjaska V, Post RM, Rogawski MA, Li H (2001). Input-specific LTP and depotentiation in the basolateral amygdala. Neuroreport 12: 635–640.
Asan E (1993). Comparative single and double immunolabelling with antisera against catecholamine biosynthetic enzymes: criteria for the identification of dopaminergic, noradrenergic and adrenergic structures in selected rat brain areas. Histochemistry 99: 427–442.
Asnis GM, Halbreich U, Rabinovich H, Ryan ND, Sachar EJ, Nelson B et al (1985). The cortisol response to desipramine in endogenous depressives and normal controls: preliminary findings. Psychiatry Res 14: 225–233.
Asnis GM, Sanderson WC, van Praag HM (1992). Cortisol response to intramuscular desipramine in patients with major depression and normal control subjects: a replication study. Psychiatry Res 44: 237–250.
Avoli M, D'Antuono M, Louvel J, Kohling R, Biagini G, Pumain R et al (2002). Network and pharmacological mechanisms leading to epileptiform synchronization in the limbic system in vitro. Prog Neurobiol 68: 167–207.
Baird AA, Gruber SA, Fein DA, Maas LC, Steingard RJ, Renshaw PF et al (1999). Functional magnetic resonance imaging of facial affect recognition in children and adolescents. J Am Acad Child Adolesc Psychiatry 38: 195–199.
Bennett BD, Huguenard JR, Prince DA (1997). Adrenoceptor-mediated elevation of ambient GABA levels activates presynaptic GABA(B) receptors in rat sensorimotor cortex. J Neurophysiol 78: 561–566.
Bennett BD, Huguenard JR, Prince DA (1998). Adrenergic modulation of GABAA receptor-mediated inhibition in rat sensorimotor cortex. J Neurophysiol 79: 937–946.
Bergles DE, Doze VA, Madison DV, Smith SJ (1996). Excitatory actions of norepinephrine on multiple classes of hippocampal CA1 interneurons. J Neurosci 16: 572–585.
Berts A, Zhong H, Minneman KP (1999). No role for Ca++ or protein kinase C in alpha-1A adrenergic receptor activation of mitogen-activated protein kinase pathways in transfected PC12 cells. Mol Pharmacol 55: 296–303.
Braga MF, Aroniadou-Anderjaska V, Xie J, Li H (2003). Bidirectional modulation of GABA release by presynaptic glutamate receptor 5 kainate receptors in the basolateral amygdala. J Neurosci 23: 442–452.
Breiter HC, Etcoff NL, Whalen PJ, Kennedy WA, Rauch SL, Buckner RL et al (1996). Response and habituation of the human amygdala during visual processing of facial expression. Neuron 17: 875–887.
Bremner JD, Innis RB, Southwick SM, Staib L, Zoghbi S, Charney DS (2000). Decreased benzodiazepine receptor binding in prefrontal cortex in combat-related posttraumatic stress disorder. Am J Psychiatry 157: 1120–1126.
Bremner JD, Krystal JH, Southwick SM, Charney DS (1996). Noradrenergic mechanisms in stress and anxiety: I. Preclinical studies. Synapse 23: 28–38.
Buchel C, Dolan RJ (2000). Classical fear conditioning in functional neuroimaging. Curr Opin Neurobiol 10: 219–223.
Buchel C, Morris J, Dolan RJ, Friston KJ (1998). Brain systems mediating aversive conditioning: an event-related fMRI study. Neuron 20: 947–957.
Cahill L, McGaugh JL (1998). Mechanisms of emotional arousal and lasting declarative memory. Trends Neurosci 21: 294–299.
Chalothorn D, McCune DF, Edelmann SE, Garcia-Cazarin ML, Tsujimoto G, Piascik MT (2002). Differences in the cellular localization and agonist-mediated internalization properties of the alpha(1)-adrenoceptor subtypes. Mol Pharmacol 61: 1008–1016.
Chen A, Hough CJ, Li H (2003). Serotonin type II receptor activation facilitates synaptic plasticity via N-Methyl-D-aspartate-mediated mechanism in the rat basolateral amygdala. Neurosci 119: 53–63.
Chen G, Ensor GR, Bohner B (1954). A facilitation action of reserpine on the central nervous system. Proc Soc Exp Biol Med 86: 507–510.
Clayton EC, Williams CL (2000). Adrenergic activation of the nucleus tractus solitarius potentiates amygdala norepinephrine release and enhances retention performance in emotionally arousing and spatial memory tasks. Behav Brain Res 112: 151–158.
Cohen RI, Almazan G (1993). Norepinephrine-stimulated PI hydrolysis in oligodendrocytes is mediated by alpha 1A-adrenoceptors. Neuroreport 4: 1115–1118.
Crow TJ, Cross AJ, Cooper SJ, Deakin JF, Ferrier IN, Johnson JA et al (1984). Neurotransmitter receptors and monoamine metabolites in the brains of patients with Alzheimer-type dementia and depression, and suicides. Neuropharmacology 23: 1561–1569.
Davidson RJ, Abercrombie H, Nitschke JB, Putnam K (1999). Regional brain function, emotion and disorders of emotion. Curr Opin Neurobiol 9: 228–234.
Davidson RJ, Slagter HA (2000). Probing emotion in the developing brain: functional neuroimaging in the assessment of the neural substrates of emotion in normal and disordered children and adolescents. Ment Retard Dev Disabil Res Rev 6: 166–170.
Davis M (1992). The role of the amygdala in fear and anxiety. Annu Rev Neurosci 15: 353–375.
Davis M (1994). The role of the amygdala in emotional learning. Int Rev Neurobiol 36: 225–266.
Davis M, Rainnie D, Cassell M (1994). Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci 17: 208–214.
Day HE, Campeau S, Watson Jr SJ, Akil H (1997). Distribution of alpha 1a-, alpha 1b- and alpha 1d-adrenergic receptor mRNA in the rat brain and spinal cord. J Chem Neuroanat 13: 115–139.
Deng XF, Chemtob S, Varma DR (1996). Characterization of alpha 1 D-adrenoceptor subtype in rat myocardium, aorta and other tissues. Br J Pharmacol 119: 269–276.
Domyancic AV, Morilak DA (1997). Distribution of alpha1A adrenergic receptor mRNA in the rat brain visualized by in situ hybridization. J Comp Neurol 386: 358–378.
Doze VA, Cohen GA, Madison DV (1991). Synaptic localization of adrenergic disinhibition in the rat hippocampus. Neuron 6: 889–900.
Drevets WC (1999). Prefrontal cortical-amygdalar metabolism in major depression. Ann NY Acad Sci 877: 614–637.
Ferry B, Magistretti PJ, Pralong E (1997). Noradrenaline modulates glutamate-mediated neurotransmission in the rat basolateral amygdala in vitro. Eur J Neurosci 9: 1356–1364.
Ferry B, Roozendaal B, McGaugh JL (1999). Basolateral amygdala noradrenergic influences on memory storage are mediated by an interaction between beta- and alpha1-adrenoceptors. J Neurosci 19: 5119–5123.
Frucht MM, Quigg M, Schwaner C, Fountain NB (2000). Distribution of seizure precipitants among epilepsy syndromes. Epilepsia 41: 1534–1539.
Fuxe K, Harfstrand A, Agnati LF, Yu ZY, Cintra A, Wikstrom AC et al (1985). Immunocytochemical studies on the localization of glucocorticoid receptor immunoreactive nerve cells in the lower brain stem and spinal cord of the male rat using a monoclonal antibody against rat liver glucocorticoid receptor. Neurosci Lett 60: 1–6.
Galvez R, Mesches MH, McGaugh JL (1996). Norepinephrine release in the amygdala in response to footshock stimulation. Neurobiol Learn Mem 66: 253–257.
Gellman RL, Aghajanian GK (1993). Pyramidal cells in piriform cortex receive a convergence of inputs from monoamine activated GABAergic interneurons. Brain Res 600: 63–73.
Gloor P (1992). Role of the amygdala in temporal lobe epilepsy. In: Aggleton JP (ed). The Amygdala: Neurobiological Aspects of Emotion, Memory, and Mental Dysfunction. Wiley-Liss Incorporation: New York. pp 505–538.
Goldstein LE, Rasmusson AM, Bunney BS, Roth RH (1996). Role of the amygdala in the coordination of behavioral, neuroendocrine, and prefrontal cortical monoamine responses to psychological stress in the rat. J Neurosci 16: 4787–4798.
Gross-Isseroff R, Dillon KA, Fieldust SJ, Biegon A (1990). Autoradiographic analysis of alpha 1-noradrenergic receptors in the human brain postmortem. Effect of suicide. Arch Gen Psychiatry 47: 1049–1053.
Habib KE, Gold PW, Chrousos GP (2001). Neuroendocrinology of stress. Endocrinol Metab Clin North Am 30: 695–728.
Joels M, de Kloet ER (1989). Effects of glucocorticoids and norepinephrine on the excitability in the hippocampus. Science 245: 1502–1505.
Kawaguchi Y, Shindou T (1998). Noradrenergic excitation and inhibition of GABAergic cell types in rat frontal cortex. J Neurosci 18: 6963–6976.
Khorchid A, Cui Q, Molina-Holgado E, Almazan G (2002). Developmental regulation of alpha 1A-adrenoceptor function in rat brain oligodendrocyte cultures. Neuropharmacology 42: 685–696.
Knepper SM, Buckner SA, Brune ME, deBernardis JF, Meyer MD, Hancock AA (1995). A-61603, a potent alpha 1-adrenergic receptor agonist, selective for the alpha 1A receptor subtype. J Pharmacol Exp Ther 274: 97–103.
Kulik A, Haentzsch A, Luckermann M, Reichelt W, Ballanyi K (1999). Neuron-glia signaling via alpha(1) adrenoceptor-mediated Ca(2+) release in Bergmann glial cells in situ. J Neurosci 19: 8401–8408.
LaBar KS, Gatenby JC, Gore JC, LeDoux JE, Phelps EA (1998). Human amygdala activation during conditioned fear acquisition and extinction: a mixed-trial fMRI study. Neuron 20: 937–945.
LeDoux JE (1992). Brain mechanisms of emotion and emotional learning. Curr Opin Neurobiol 2: 191–197.
Lepretre N, Mironneau J, Morel JL (1994). Both alpha 1A- and alpha 2A-adrenoreceptor subtypes stimulate voltage- operated L-type calcium channels in rat portal vein myocytes. Evidence for two distinct transduction pathways. J Biol Chem 269: 29546–29552.
Li H, Chen A, Xing G, Wei ML, Rogawski MA (2001). Kainate receptor-mediated heterosynaptic facilitation in the amygdala. Nat Neurosci 4: 612–620.
Li R, Nishijo H, Ono T, Ohtani Y, Ohtani O (2002). Synapses on GABAergic neurons in the basolateral nucleus of the rat amygdala: double-labeling immunoelectron microscopy. Synapse 43: 42–50.
Lippa AS, Klepner CA, Yunger L, Sano MC, Smith WV, Beer B (1978). Relationship between benzodiazepine receptors and experimental anxiety in rats. Pharmacol Biochem Behav 9: 853–856.
Madison DV, Nicoll RA (1988). Norepinephrine decreases synaptic inhibition in the rat hippocampus. Brain Res 442: 131–138.
McEwen BS (2002). Protective and damaging effects of stress mediators: the good and bad sides of the response to stress. Metabolism 51: 2–4.
McKernan MG, Shinnick-Gallagher P (1997). Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390: 607–611.
Medina JH, Novas ML, De Robertis E (1983). Changes in benzodiazepine receptors by acute stress: different effect of chronic diazepam or RO 15-1788 treatment. Eur J Pharmacol 96: 181–185.
Miller LG, Thompson ML, Greenblatt DJ, Deutsch SI, Shader RI, Paul SM (1987). Rapid increase in brain benzodiazepine receptor binding following defeat stress in mice. Brain Res 414: 395–400.
Miyahara S, Komori T, Fujiwara R, Shizuya K, Yamamoto M, Ohmori M et al (1999). Effects of single and repeated stresses on the expression of mRNA for alpha1-adrenoceptors in the rat hypothalamus and midbrain. Eur J Pharmacol 379: 111–114.
Ottenweller JE, Natelson BH, Pitman DL, Drastal SD (1989). Adrenocortical and behavioral responses to repeated stressors: toward an animal model of chronic stress and stress-related mental illness. Biol Psychiatry 26: 829–841.
Papadopoulos GC, Parnavelas JG (1990). Distribution and synaptic organization of serotoninergic and noradrenergic axons in the lateral geniculate nucleus of the rat. J Comp Neurol 294: 345–355.
Pawlak R, Magarinos AM, Melchor J, McEwen B, Strickland S (2003). Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat Neurosci 6: 168–174.
Perez DM, DeYoung MB, Graham RM (1993). Coupling of expressed alpha 1B- and alpha 1D-adrenergic receptor to multiple signaling pathways is both G protein and cell type specific. Mol Pharmacol 44: 784–795.
Pitkänen A (2000). Connectivity of the rat amygdaloid complex. In: Aggleton JP (ed.) The Amygdala: A Functional Analysis. Oxford University Press: Oxford, UK. pp 31–99.
Pitkanen A, Pikkarainen M, Nurminen N, Ylinen A (2000). Reciprocal connections between the amygdala and the hippocampal formation, perirhinal cortex, and postrhinal cortex in rat. A review. Ann NY Acad Sci 911: 369–391.
Quirarte GL, Galvez R, Roozendaal B, McGaugh JL (1998). Norepinephrine release in the amygdala in response to footshock and opioid peptidergic drugs. Brain Res 808: 134–140.
Rauch SL, Whalen PJ, Shin LM, McInerney SC, Macklin ML, Lasko NB et al (2000). Exaggerated amygdala response to masked facial stimuli in posttraumatic stress disorder: a functional MRI study. Biol Psychiatry 47: 769–776.
Rogan MT, Staubli UV, LeDoux JE (1997). Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390: 604–607.
Saussy Jr DL, Goetz AS, Queen KL, King HK, Lutz MW, Rimele TJ (1996). Structure activity relationships of a series of buspirone analogs at alpha-1 adrenoceptors: further evidence that rat aorta alpha-1 adrenoceptors are of the alpha-1D-subtype. J Pharmacol Exp Ther 278: 136–144.
Schneider F, Grodd W, Weiss U, Klose U, Mayer KR, Nagele T et al (1997). Functional MRI reveals left amygdala activation during emotion. Psychiatry Res 76: 75–82.
Schoepp DD, Rutledge CO (1985). Comparison of postnatal changes in alpha 1-adrenoceptor binding and adrenergic stimulation of phosphoinositide hydrolysis in rat cerebral cortex. Biochem Pharmacol 34: 2705–2711.
Schwinn DA, Page SO, Middleton JP, Lorenz W, Liggett SB, Yamamoto K et al (1991). The alpha 1C-adrenergic receptor: characterization of signal transduction pathways and mammalian tissue heterogeneity. Mol Pharmacol 40: 619–626.
Seguela P, Watkins KC, Geffard M, Descarries L (1990). Noradrenaline axon terminals in adult rat neocortex: an immunocytochemical analysis in serial thin sections. Neuroscience 35: 249–264.
Seligman ME, Beagley G (1975). Learned helplessness in the rat. J Comp Physiol Psychol 88: 534–541.
Seligman ME, Maier SF (1967). Failure to escape traumatic shock. J Exp Psychol 74: 1–9.
Servatius RJ, Ottenweller JE, Natelson BH (1995). Delayed startle sensitization distinguishes rats exposed to one or three stress sessions: further evidence toward an animal model of PTSD. Biol Psychiatry 38: 539–546.
Stanford SC (1995). Central noradrenergic neurones and stress. Pharmacol Ther 68: 242–297.
Stanton PK (1992). Noradrenergic modulation of epileptiform bursting and synaptic plasticity in the dentate gyrus. Epilepsy Res 7(Suppl): 135–150.
Stanton PK, Mody I, Zigmond D, Sejnowski T, Heinemann U (1992). Noradrenergic modulation of excitability in acute and chronic model epilepsies. Epilepsy Res 8(Suppl): 321–334.
Stone EA, McEwen BS, Herrera AS, Carr KD (1987). Regulation of alpha and beta components of noradrenergic cyclic AMP response in cortical slices. Eur J Pharmacol 141: 347–356.
Stone EA, Platt JE, Herrera AS, Kirk KL (1986). Effect of repeated restraint stress, desmethylimipramine or adrenocorticotropin on the alpha and beta adrenergic components of the cyclic AMP response to norepinephrine in rat brain slices. J Pharmacol Exp Ther 237: 702–707.
Stone EA, Quartermain D (1999). Alpha-1-noradrenergic neurotransmission, corticosterone, and behavioral depression. Biol Psychiatry 46: 1287–1300.
Stoop R, Epiney S, Meier E, Pralong E (2000). Modulation of epileptiform discharges in the rat limbic system in vitro by noradrenergic agents. Neurosci Lett 287: 5–8.
Strakowski SM, DelBello MP, Sax KW, Zimmerman ME, Shear PK, Hawkins JM et al (1999). Brain magnetic resonance imaging of structural abnormalities in bipolar disorder. Arch Gen Psychiatry 56: 254–260.
Szot P, Weinshenker D, White SS, Robbins CA, Rust NC, Schwartzkroin PA et al (1999). Norepinephrine-deficient mice have increased susceptibility to seizure-inducing stimuli. J Neurosci 19: 10985–10992.
Tanaka M, Yoshida M, Emoto H, Ishii H (2000). Noradrenaline systems in the hypothalamus, amygdala and locus coeruleus are involved in the provocation of anxiety: basic studies. Eur J Pharmacol 405: 397–406.
Temkin NR, Davis GR (1984). Stress as a risk factor for seizures among adults with epilepsy. Epilepsia 25: 450–456.
Vanitallie TB (2002). Stress: a risk factor for serious illness. Metabolism 51: 40–45.
Vermetten E, Bremner JD (2002). Circuits and systems in stress. I. Preclinical studies. Depress Anxiety 15: 126–147.
Villarreal G, King CY (2001). Brain imaging in posttraumatic stress disorder. Semin Clin Neuropsychiatry 6: 131–145.
Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S (2002). Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci 22: 6810–6818.
Weinshenker D, Szot P, Miller NS, Rust NC, Hohmann JG, Pyati U et al (2001). Genetic comparison of seizure control by norepinephrine and neuropeptide Y. J Neurosci 21: 7764–7769.
Weiss SRB, Li H, Sitcoske-O'Shea M, Post RM (2000). Amygdala plasticity: the neurobiological implications of kindling. In: Aggleton JP (ed.) The Amygdala: A Functional Analysis. Oxford University Press: Oxford, UK. pp 31–99.
Weizman R, Weizman A, Kook KA, Vocci F, Deutsch SI, Paul SM (1989). Repeated swim stress alters brain benzodiazepine receptors measured in vivo. J Pharmacol Exp Ther 249: 701–707.
Whalen PJ, Rauch SL, Etcoff NL, McInerney SC, Lee MB, Jenike MA (1998). Masked presentations of emotional facial expressions modulate amygdala activity without explicit knowledge. J Neurosci 18: 411–418.
Williams AM, Nguyen ML, Morilak DA (1997). Co-localization of alpha1D adrenergic receptor mRNA with mineralocorticoid and glucocorticoid receptor mRNA in rat hippocampus. J Neuroendocrinol 9: 113–119.
Williams CL, Men D, Clayton EC (2000). The effects of noradrenergic activation of the nucleus tractus solitarius on memory and in potentiating norepinephrine release in the amygdala. Behav Neurosci 114: 1131–1144.
Wu D, Katz A, Lee CH, Simon MI (1992). Activation of phospholipase C by alpha 1-adrenergic receptors is mediated by the alpha subunits of Gq family. J Biol Chem 267: 25798–25802.
Xiao L, Jeffries WB (1998). Kinetics of alkylation of cloned rat alpha1-adrenoceptor subtypes by chloroethylclonidine. Eur J Pharmacol 347: 319–327.
Yang M, Ruan J, Voller M, Schalken J, Michel MC (1999). Differential regulation of human alpha1-adrenoceptor subtypes. Naunyn Schmiedebergs Arch Pharmacol 359: 439–446.
Zhong H, Minneman KP (1999). Alpha1-adrenoceptor subtypes. Eur J Pharmacol 375: 261–276.
Acknowledgements
We thank Dr Robert M Post for critical review of the manuscript and valuable discussions. The expert assistance of Eleanore Gamble and Dr Jozsef Czeee is greatly appreciated. CJH and VA-A were financially supported by the Stanley Foundation. This work was supported in part by DAMD Grant 17-00-1-0110 and USUHS Grant RO88DC to HL.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Braga, M., Aroniadou-Anderjaska, V., Manion, S. et al. Stress Impairs α1A Adrenoceptor-Mediated Noradrenergic Facilitation of GABAergic Transmission in the Basolateral Amygdala. Neuropsychopharmacol 29, 45–58 (2004). https://doi.org/10.1038/sj.npp.1300297
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300297
Keywords
This article is cited by
-
Noradrenergic stimulation of α1 adrenoceptors in the medial prefrontal cortex mediates acute stress-induced facilitation of seizures in mice
Scientific Reports (2023)
-
Aversive Stress Reduces Mu Opioid Receptor Expression in the Intercalated Nuclei of the Rat Amygdala
Cellular and Molecular Neurobiology (2021)
-
Stress induces insertion of calcium-permeable AMPA receptors in the OFC–BLA synapse and modulates emotional behaviours in mice
Translational Psychiatry (2020)
-
Prazosin during fear conditioning facilitates subsequent extinction in male C57Bl/6N mice
Psychopharmacology (2019)
-
The Role of β1,2-Adrenoceptors in the Amygdala in the Behavior of Rats with Different Levels of Freezing in Conditioned Reflex Fear
Neuroscience and Behavioral Physiology (2019)
