
Abstract
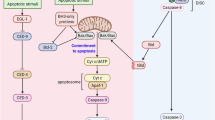
There has been a considerable debate as to whether caspase-2 is an initiator or effector caspase. Recently, a new model of intrinsic pathway of apoptosis has been proposed, which suggests caspase-2 to be an initiator caspase. For example, ultraviolet radiation (UV) and other DNA damage-inducing agents were shown to first activate caspase-2 and then regulate the mitochondrial and postmitochondrial events. Active caspase-2 was found to engage mitochondria by promoting Bax translocation to the mitochondria. Consequently, Bax was proposed to play a central role in bridging the active caspase-2 with mitochondria by affecting mitochondrial permeability, cytochrome c release into the cytosol and caspase-9 activation. In the present study, we investigated the role of Bax in UV-induced apoptosis and caspase-2 activation. Our results indicate that UV-induced apoptosis and caspase-2 activation were diminished in Bax-deficient cells, suggesting that Bax appears to play an important role in UV-induced apoptosis as well as caspase-2 activation, and that it also appears to reside upstream of caspase-2. Bax deficiency also affected the activation of caspase-3 and -8 and abolished caspase-9 activation during UV-induced apoptosis, suggesting that the absence of caspase-9 activation may affect caspase-2, -3 and -8 activation in Bax-deficient cells. Based on our results, we propose that activation of caspases is not a linear cascade of events, but is rather connected via complex feedback loops.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others
References
Aragane Y, Klums D, Metze D, Wikes G, Poppelmann B, Luger TA and Schwarz T . (1998). J. Cell Biol., 140, 171–182.
Ashkenazi A and Dixit VM . (1999). Curr. Opin. Cell Biol., 11, 255–260.
Cowling V and Downward J . (2002). Cell Death Differ., 9, 1046–1056.
Fujita E, Egashira J, Urase K, Kuida K and Momoi T . (2001). Cell Death Differ., 8, 335–344.
Gross A, McDonnell JM and Korsmeyer SJ . (2000). Genes Dev., 13, 1899–1911.
Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T and Alnemri ES . (2002). J. Biol. Chem., 277, 13430–134307.
Harvey NL, Butt AJ and Kumar S . (1997). J. Biol. Chem., 272, 13134–13139.
He Q, Luo X, Huang Y and Sheikh MS . (2002). Oncogene, 21, 6032–6040.
Hengartner MO . (2000). Nature, 407, 770–776.
Holcik M and Korneluk RG . (2001). Nat. Rev. Mol. Cell Biol., 2, 550–556.
Huang Y, He Q, Hillman MJ, Rong R and Sheikh MS . (2001). Cancer Res., 61, 6918–6924.
Lassus P, Opitz-Araya X and Lazebnik Y . (2002). Science, 297, 1352–1354.
Li H, Bergeron L, Cryns V, Pasternack MS, Zhu H, Shi L, Greenberg A and Yuan J . (1997). J. Biol. Chem., 272, 21010–21017.
Paroni G, Henderson C, Schneider C and Brancolini C . (2001). J. Biol. Chem., 276, 21907–21915.
Rehemtulla A, Hamilton CA, Chinnaiyan AM and Dixit VM . (1997). J. Biol. Chem., 272, 25783–257836.
Robertson JD, Enoksson M, Suomela M, Zhivotovsky B and Orrenius S . (2002). J. Biol. Chem., 2002277, 29803–29809.
Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S and Peter ME . (1998). Eur. J. Biochem., 254, 439–459.
Sheikh MS, Antinore MJ, Huang Y and Fornace Jr AJ . (1998). Oncogene, 17, 2555–2563.
Sheikh MS and Fornace Jr AJ . (2000). J. Cell. Physiol., 182, 171–181.
Sitailo LA, Tibudan SS and Denning MF . (2002). J. Biol. Chem., 277, 19346–19352.
Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR and Martin SJ . (1999). J. Cell Biol., 144, 281–292.
Thornberry NA and Lazebnik Y . (1998). Science, 281, 1312–1316.
Wang L, Miura M, Bergeron L, Zhu H and Yuan J . (1994). Cell., 78, 739–750.
Wang X . (2001). Genes Dev., 15, 2922–2933.
Zhang L, Yu J, Park BH, Kinzler KW and Vogelstein B . (2000). Science, 290, 989–992.
Acknowledgements
We thank Dr Bert Vogelstein (Johns Hopkins University, Baltimore, MD), for kindly providing the Bax-proficient and -deficient cells used in this study. This work was supported in part by NIH grants CA89043, CA86945 and a Department of Defense grant DMAD 170010722.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
He, Q., Huang, Y. & Sheikh, M. Bax deficiency affects caspase-2 activation during ultraviolet radiation-induced apoptosis. Oncogene 23, 1321–1325 (2004). https://doi.org/10.1038/sj.onc.1207212
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1207212
Keywords
This article is cited by
-
Translocation and oligomerization of Bax is regulated independently by activation of p38 MAPK and caspase-2 during MN9D dopaminergic neurodegeneration
Apoptosis (2011)
-
The enigma of caspase-2: the laymen's view
Cell Death & Differentiation (2009)
-
Catalase, Bax and p53 expression in the visual system of the crab Ucides cordatus following exposure to ultraviolet radiation
Cell and Tissue Research (2007)
-
Tumor necrosis factor α sensitizes malignant cells to chemotherapeutic drugs via the mitochondrial apoptosis pathway independently of caspase-8 and NF-κB
Oncogene (2004)
