
Abstract
Aim:
Sphingosine kinase 1 (SPHK1) is involved in various cellular functions, including cell growth, migration, apoptosis, cytoskeleton architecture and calcium homoeostasis, etc. As an oncogenic kinase, SPHK1 is associated with the development and progression of cancers. The aim of this study was to investigate whether SPHK1 was involved in hepatocarcinogenesis induced by the hepatitis B virus X protein (HBx).
Methods:
The expression of SPHK1 in hepatocellular carcinoma (HCC) tissue and hepatoma cells were measured using qRT-PCR and Western blot analysis. HBx expression levels in hepatoma cells were modulated by transiently transfected with HBx or psi-HBx plasmids. The SPHK1 promoter activity was measured using luciferase reporter gene assay, and the interaction of the transcription factor AP2α with the SPHK1 promoter was studied with chromatin immunoprecipitation assay. The growth of hepatoma cells was evaluated in vitro using MTT and colony formation assays, and in a tumor xenograft model.
Results:
A positive correlation was found between the mRNA levels of SPHK1 and HBx in 38 clinical HCC samples (r=+0.727, P<0.01). Moreover, the expression of SPHK1 was markedly increased in the liver cancer tissue of HBx-transgenic mice. Overexpressing HBx in normal liver cells LO2 and hepatoma cells HepG2 dose-dependently increased the expression of SPHK1, whereas silencing HBx in HBx-expressing hepatoma cells HepG2-X and HepG2.2.15 suppressed SPHK1 expression. Furthermore, overexpressing HBx in HepG2 cells dose-dependently increased the SPHK1 promoter activity, whereas silencing HBx in HepG2-X cells suppressed this activity. In HepG2-X cells, AP2α was found to directly interact with the SPHK1 promoter, and silencing AP2α suppressed the SPHK1 promoter activity and SPHK1 expression. Silencing HBx in HepG2-X cells abolished the HBx-enhanced proliferation and colony formation in vitro, and tumor growth in vivo.
Conclusion:
HBx upregulates SPHK1 through the transcription factor AP2α, which promotes the growth of human hepatoma cells
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors in China and the fifth most common cancer worldwide. Because of its late diagnosis and high recurrence rate, HCC is an aggressive human cancer and the second leading cause of death in the world1. Chronic infection with the hepatitis B virus (HBV) has been directly linked with the development of HCC. The HBV X protein (HBx), encoded by the HBx gene, has been detected with a high frequency in liver cells from patients suffering from chronic hepatitis, cirrhosis, and liver cancer2,3. Extensive evidence shows that HBx is a multifunctional regulatory protein and plays a crucial role in hepatocarcinogenesis, which involves modulation of cell growth regulatory genes, regulation of apoptosis and inhibition of nucleotide excision cellular DNA4,5,6. Our group has reported that HBx upregulates oncoproteins Rab18, Lin28A/Lin28B and YAP and modulates non-coding RNA HULC and miR-205 to promote hepatocarcinogenesis7,8,9,10,11. HBx protein promotes liver cell proliferation via a positive cascade loop involving arachidonic acid metabolism and p-ERK1/212. However, the underlying mechanism of hepatocarcinogenesis mediated by HBx is not well documented.
Recent studies indicate that high-risk cancer is linked to metabolic disorders, such as those impacting lipids and sphingolipids. Sphingosine kinase 1 (SPHK1) is a conserved enzyme that generates Sphingosine-1-phosphate (S1P) to regulate neurotransmission, synaptic function and neuronal cell proliferation by activating five G-protein-coupled receptors (S1P1-S1P5). SPHK1 plays a crucial role in the regulation of the sphingolipid biostat13. Mounting evidence suggests that SPHK1 is associated with various cellular functions such as cell growth, migration, apoptosis, cytoskeleton architecture and calcium homoeostasis. As an oncogenic kinase, SPHK1 is upregulated in several types of cancers and is correlated with the development and progression of the disease. It has been reported that SPHK1/S1P is involved in chronic intestinal inflammation and colon tumorigenesis14, and SPHK1 overexpression contributes to cetuximab resistance in human colorectal cancer models15. Sphingosine kinase 1 promotes tumor cell migration and invasion via the S1P/EDG1 axis in HCC16. SPHK1 regulates proliferation and survival responses in triple-negative breast cancer17. Furthermore, inhibition of SPHK1 can suppress the migration and proliferation of ovarian cancer and hepatocellular carcinoma cells18,19,20. Given that SPHK1 is upregulated in HCC, we predict that SPHK1 might be involved in the hepatocarcinogenesis that is mediated by HBx.
In this study, we are interested in the role of HBx in the modulation of SPHK1 to enhance the growth of hepatoma cells. Interestingly, our data indicate that HBx is able to elevate the expression of SPHK1 in hepatoma cells by upregulating transcription factor AP2α. Our findings provide new insights into the mechanism by which HBx enhances proliferation of hepatoma cells.
Materials and methods
Patient and HBx-transgenic (HBx-Tg) mice samples
Thirty-eight HCC tissue samples utilized in this study were obtained from Tianjin Tumor Hospital (Tianjin, China) after surgical resection. Clinical information regarding patients was obtained from patient records and is summarized in Supplementary Table 1. All patients were diagnosed with primary HCC, and none had received previous radiotherapy or chemotherapy before surgery. Written consent approving the use of their tissues for research purposes was obtained from each patient following the operation. The liver and HCC tissues of HBx-Tg mice were obtained from the Genetic Laboratory of Development and Diseases, Institute of Biotechnology (Beijing, China)21. The study was approved by the Institute Research Ethics Committee at Nankai University (Tianjin, China).
Cell lines and cell culture
Hepatoma cell lines HepG2, HepG2-P (a HepG2 cell line with an integrated pcDNA3.1 vector), HepG2-X (a HepG2 cell line with an integrated pcDNA3.1-HBx vector) and HepG2.2.15 (a HepG2 cell line with integrated full-length HBV DNA) were maintained in Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY, USA)9. Liver cell lines LO2, LO2-P (a LO2 cell line with an integrated pcDNA3.1 vector), and LO2-X (a LO2 cell line with an integrated pcDNA3.1-HBx vector) were cultured in RPMI-1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal calf serum (FCS), 100 U/mL penicillin, and 100 mg/mL streptomycin in 5% CO2 at 37 °C22.
RNA extraction, reverse-transcription and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from cells (or tumor tissues) using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The total extraction was treated with DNase I (Sigma, St Louis, MO, USA). First-strand cDNA was synthesized as previously reported23. Reverse transcription was performed using poly(A)-tailed total RNA and reverse transcription primer with ImPro-II Reverse Transcriptase (Promega, Madison, WI, USA) according to the manufacturer's protocol. qRT-PCR was performed using a Bio-Rad sequence detection system according to the manufacturer's instructions using double-stranded, DNA-specific Fast Start Universal SYBR Green Master (Roche, Indianapolis, IN, USA). Experiments were conducted in duplicate in three independent assays. Relative transcriptional folds were calculated as 2-ΔΔCt. GAPDH was used as an internal control for normalization. All of the primers used in this study are listed in Supplementary Table 2.
Western blot analysis
Western blot analysis was carried out using a previously described protocol10. The following primary antibodies were used: mouse anti-HBx (Abcam, Cambridge, UK), rabbit anti-SPHK1 (Proteintech, Wuhan, China), rabbit anti-AP2α (Proteintech), mouse anti-β-actin (NeoMarkers, MA, USA), and rabbit anti-β-actin (NeoMarkers, MA, USA).
RNA interfering (RNAi)
The short interfering RNA (siRNA) duplexes targeting AP2α (si-h-TFAP2A_001, si-h-TFAP2A_002, si-h-TFAP2A_003) were designed and synthesized by Ribobio Co Lit (Guangzhou, China). The sequences of si-SPHK1 and the construction of psi-HBx were reported in previous studies9,24. The control cells were treated with 100 nmol/L si-Control (NControl_05815) synthesized by Ribobio Co Lit or 2 μg pSilence vector. The dose of siRNA and si-Control was determined according to the product specification from Ribobio Co Lit.
Luciferase reporter gene assays
Luciferase reporter gene assays were performed using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer's instructions. Cells were transferred into 24-well plates at 3×104 cells per well. After 24 h, the cells were transiently co-transfected with 0.1 μg/well of pRL-TK plasmid (Promega) containing the Renilla luciferase gene used for internal normalization and constructs containing the core region of the SPHK1 promoter (the wild type pGL3-320 and the mutant pGL3-320-mut). The luciferase activities were measured as previously described9,12. All experiments were performed at least three times.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed in HepG2-X cells, as previously reported9. The promoter region of the SPHK1 that included the AP2α binding site was amplified from the immunoprecipitated DNA samples with specific primers. All primers are listed in Supplementary Table 2.
MTT assays
HepG2 cells were seeded onto 96-well plates (1000 cells/well) for 24 h before transfection, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays were used to assess cell proliferation daily from the first day until the third day after transfection. The MTT protocol was previously described25.
Analysis of colony formation
For clonogenicity analysis, 48 h after transfection, 1000 viable transfected cells (LO2-P, LO2-X, LO2-X+si-SPHK1; HepG2-P, HepG2-X, HepG2-X+si-SPHK1) were placed in 6-well plates and maintained in complete medium for 2 weeks. Colonies were fixed with methanol and stained with methylene blue.
Animal transplantation
Nude mice were housed and treated according to the guidelines established by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. We conducted the animal transplantation according to the Declaration of Helsinki. Tumor transplantation in nude mice was performed as follows: cells were harvested and re-suspended at 2×107 per mL with sterile phosphate-buffered saline. Groups of 4-week-old female BALB/c athymic nude mice (Experiment Animal Center of Peking, China) (each group, n=5) were subcutaneously injected at the shoulder with 0.2 mL of the cell suspensions. Tumor growth was measured after 10 days post-injection and subsequently every 5 d. Tumor volume (V) was monitored by measuring the length (L) and width (W) of the tumor with calipers and calculated with the formula (L×W2)×0.5. After 30 d, tumor-bearing mice and controls were euthanized, and the tumors were excised and measured.
Statistical analysis
Each experiment was repeated a minimum of three times. Statistical significance was assessed by comparing mean values (±standard deviation, SD) using Student's t-test for independent groups, and significance was assumed for P<0.05 and P<0.01. Pearson's correlation coefficient was used to determine the correlation between the levels of HBx and SPHK1 mRNA in HCC tissues.
Results
The mRNA levels of HBx are positively associated with those of SPHK1 in clinical HCC tissues
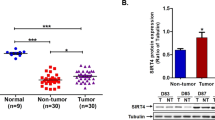
It has been reported that the mRNA levels of SPHK1 are upregulated in liver cancer tissues16. We evaluated the relationship between HBx and SPHK1 in clinical HCC tissues. Our data showed that the mRNA levels of HBx were positively correlated with those of SPHK1 in 38 clinical HCC samples (P<0.01, r=0.727, Pearson's correlation) (Figure 1A). Further, we detected the expression of SPHK1 in the liver or HCC tissues from HBx-Tg mice. Interestingly, we found that SPHK1 was upregulated in 6-month-old HBx-Tg mice (Figure 1B). SPHK1 was especially highly expressed in liver cancer tissues from HBx-Tg mice aged 18 months, suggesting that HBx is able to upregulate SPHK1 in HBx-Tg mice. With these two findings, we conclude that the mRNA levels of HBx are positively associated with those of SPHK1 in clinical HCC tissues.

The mRNA levels of HBx are positively associated with those of SPHK1 in clinical HCC tissues. (A) The correlation between HBx mRNA levels and SPHK1 mRNA levels was detected by qRT-PCR in clinical HCC tissues (P<0.01, r=0.727, Pearson's correlation). (B) The expression of SPHK1 at the levels of mRNA and protein was examined using RT-PCR and Western blot analysis in either the liver tissues or the HCC tissues of HBx-Tg mice.
HBx is able to upregulate the expression of SPHK1
To further validate the relationship between HBx and SPHK1, we examined the expression of SPHK1 in LO2 or HepG2 cells transiently transfected with pcDNA3.1-HBx using RT-PCR and Western blot analysis. We found that HBx could upregulate SPHK1 in a dose-dependent manner at both the levels of mRNA and protein (Figure 2A and 2C). We also observed that SPHK1 was upregulated in HepG2-X and HepG2.2.15 cell lines (Supplementary Figure 1). In contrast, depletion of HBx by psi-HBx could downregulate the expression of SPHK1 in HepG2-X and HepG2.2.15 cells in a dose-dependent manner at both the levels of mRNA and protein (Figure 2B and 2D), suggesting that HBx is able to upregulate SPHK1 in hepatoma cells. Thus, we conclude that HBx upregulates the expression of SPHK1 in hepatoma cells.

HBx is able to upregulate the expression of SPHK1. (A, B) The mRNA expression of SPHK1 was examined by RT-PCR in LO2 and HepG2 (or HepG2-X and HepG2.2.15) cells that were transfected with the pcDNA3.1-HBx plasmid (or psi-HBx). The transfection efficiency of HBx (or psi-HBx) was detected by RT-PCR. (C, D) The expression of SPHK1 was examined using Western blot analysis in LO2 and HepG2 (or HepG2-X and HepG2.2.15) cells that were transfected with the pcDNA3.1-HBx plasmid (or psi-HBx). The transfection efficiency of HBx (or psi-HBx) was detected using Western blot analysis of the cells.
HBx activates SPHK1 promoter through upregulating transcription factor AP2α
To determine the underlying mechanism by which HBx upregulates SPHK1, we investigated the transcriptional regulation of SPHK1. We found that HBx was able to enhance the activity of the SPHK1 promoter in HepG2 cells in a dose-dependent manner (Figure 3A), but HBx RNA interference decreased the activity of the SPHK1 promoter in HepG2-X cells (Figure 3B), suggesting that HBx activates the transcription of SPHK1. We then analyzed the promoter of SPHK1 using a bioinformatic tool (http://alggen.lsi.upc.edu). Interestingly, we observed an AP2α binding site in the -20 to -15 region of the SPHK1 promoter. AP2α plays an important role in the development of cancers. It has been reported that HBx upregulates AP2α in hepatoma cells to regulate Raf1 promoter activity26. We further validated the expression levels of AP2α by Western blot analysis in HBx-overexpressed HepG2 and HepG2.2.15 cell lines (Supplementary Figure 2A and 2B), supporting the idea that HBx is able to upregulate AP2α in hepatoma cells. Therefore, we investigated the role of AP2α in activation of the SPHK1 promoter mediated by HBx. We constructed a mutant version of the SPHK1 promoter, termed pGL3-320-mut, with a deletion of six nucleotides in the binding site of AP2α (Figure 3C). As expected, we observed that the relative luciferase activity of pGL3-320-mut was decreased compared to that of pGL3-320 in HepG2 and LO2 cells (Figure 3C). Moreover, we measured the expression levels of SPHK1 at the mRNA and protein levels using RT-PCR and Western blot analysis, respectively, in HepG2-X cells transiently transfected with si-AP2α. We found that AP2α siRNA could downregulate the expression of SPHK1 in HepG2-X in a dose-dependent manner (Figure 3D and Supplementary Figure 2C), suggesting that AP2α is required for the transcription of SPHK1. ChIP assay further showed that AP2α was able to directly interact with the SPHK1 promoter in HepG2-X cells (Figure 3E). In addition, an AP2α siRNA pool significantly abolished the HBx-increased activation of the SPHK1 promoter (Figure 3F), suggesting that AP2α is responsible for the activation of the SPHK1 promoter increased by HBx. Interference efficiency of the si-AP2α pool was detected in HepG2 cells by Western blot analysis (Figure 3G). These two findings indicate that HBx activates the SPHK1 promoter by upregulating transcription factor AP2α in hepatoma cells.

HBx activates the SPHK1 promoter by upregulating the transcription factor AP2α. (A, B) The activity of the SPHK1 promoter (pGL3-320) was measured by luciferase reporter gene assays in HepG2 (or HepG2-X) cells that were co-transfected with pGL3-320 and pcDNA3.1-HBx (or psi-HBx). (C) A model demonstrates the predicted conserved AP2α binding site at nucleotides -20/-15 of the SPHK1 promoter. The generated mutant site at the SPHK1 promoter region is indicated. The luciferase activities of pGL3-320 and pGL3-320-mut were examined by luciferase reporter gene assays in HepG2 (or LO2) cells. (D) The expression of SPHK1 was detected using Western blot analysis in HepG2-X cells transfected with si-AP2α. (E) The interaction of AP2α with SPHK1 promoter was determined by ChIP assays in HepG2-X cells. (F) The relative activity of pGL3-320 was measured in HepG2 cells that were co-transfected with pcDNA3.1-HBx and si-AP2α. (G) The interfering efficiency of the siRNA pool targeting AP2α (si-AP2α) was validated by Western blot analysis in HepG2 cells. The data are shown as the mean±SD of three independent experiments. bP<0.05, cP<0.01. Student's t-test.
HBx promotes proliferation of hepatoma cells through SPHK1 in vitro and in vivo
Next, we evaluated the role of SPHK1 in promoting proliferation of hepatoma cells mediated by HBx. MTT assays showed that HBx was able to accelerate the proliferation of hepatoma cells. However, SPHK1 siRNA could block the promotion of cell proliferation mediated by HBx (Figure 4A). A colony formation assay showed that HBx increased the size and number of the colonies of LO2 and HepG2 cells. In contrast, SPHK1 siRNA blocked this event (Figure 4B), suggesting that SPHK1 is involved in the HBx-enhanced proliferation of hepatoma cells. We further verified these data in vivo using a tumor xenograft model. We observed that HBx accelerated the growth of tumors transplanted into nude mice, but the treatment with si-SPHK1 could markedly block the tumor growth (Figure 5A–5C). The expression levels of HBx and SPHK1 were confirmed by Western blot analysis in the tumor tissues of mice (Figure 5D). In addition, immunohistochemistry staining of Ki-67, a marker of proliferation, further attested that interference by SPHK1 blocked the HBx-enhanced tumor growth (Figure 5E), suggesting that SPHK1 plays a crucial role in the tumor growth mediated by HBx. With these two findings, we conclude that HBx promotes hepatoma growth through SPHK1 in vitro and in vivo.

HBx promotes proliferation of hepatoma cells through SPHK1 in vitro. (A) The effect of SPHK1 on HBx-enhanced proliferation of HepG2 cells was measured with MTT assays. (B) The effect of SPHK1 on HBx-enhanced clonogenicity of HepG2 (or LO2) cells was detected by clone formation assays. bP<0.05, cP<0.01.

HBx accelerates tumor growth through SPHK1 in vivo. (A) Photographs of tumors from nude mice (n=5) that were transplanted with HepG2-X cells pretreated with si-SPHK1 are shown. (B) The growth curves of tumors are depicted. (C) The average tumor weights from mice are depicted. (D) The protein levels of HBx and SPHK1 in the tumor tissues were examined using Western blot analysis, respectively. (E) The expression of Ki-67 in the tumor tissues of mice was detected using IHC. Statistically significant differences are indicated: cP<0.01. Student's t-test.
Discussion
HCC is one of the most common malignant cancers worldwide. Substantial evidence has suggested that HBx displays crucial functions in the pathogenesis of HCC2,27. It has been reported that SPHK1 plays an important role in the regulation of fundamental biological processes and tumorigenesis17,28,29,30. Therefore, we are interested in whether SPHK1 is involved in HBx-induced hepatocarcinogenesis.
To demonstrate the relationship between HBx and SPHK1 in HCC, we examined the mRNA levels of HBx and SPHK1 in clinical HCC tissues using qRT-PCR. Interestingly, we found that the mRNA levels of HBx were positively correlated with those of SPHK1 in the tissues. Moreover, we observed that SPHK1 was upregulated in the tissues of 6-month normal liver and 18-month liver cancer from HBx-Tg mice. This result suggests that HBx might upregulate SPHK1 in liver cancer. Our data revealed that HBx was capable of upregulating SPHK1 in hepatoma cells. SPHK1 catalyzes the phosphorylation of sphingosine to generate the bioactive lipid S1P. SPHK1 is activated by several factors, including ganglioside GM1 (GM1), G-protein coupled receptors, small GTPases, tyrosine kinase receptors, pro-inflammatory cytokines, immunoglobulin receptors, and calcium and protein kinase activators, and it is involved in numerous biological processes including cell growth, proliferation, differentiation, apoptosis, motility, and cytoskeletal rearrangement31. It has been reported that SPHK1 promotes tumor progression and confers the malignancy phenotypes of colon cancer by regulating the focal adhesion kinase pathway and adhesion molecules32. SPHK1 promotes cell proliferation, migration and invasion through the S1P/EDG1 axis and the NF-κB pathway in HCC16,19. Therefore, we hypothesizedthat SPHK1 might be involved in the hepatocarcinogenesis mediated by HBx.
Next, we further investigated the underlying mechanism by which HBx upregulates SPHK1. Interestingly, we found that HBx could activate the promoter of SPHK1. Bioinformatic analysis showed that there was an AP2α binding site in the promoter region of SPHK1. AP2α is a transcription factor that orchestrates a variety of cell processes, including cell growth and tissue differentiation33,34. It has been reported that the AP2 binding motif of the promoter region of the SPHK1 gene is important for its induction by the glial cell line-derived neurotrophic factor (GDNF)35. Overexpression of AP-2α activates the transcription activities of Hoxa7, Hoxa9 and Meis1 and regulates acute myeloid leukemia cell proliferation36. Dysregulation of AP-2α expression is associated with impaired IFN-γ actions in cancer cells37. Thus, we selected AP2α for further study. Interestingly, we showed that AP2α was required for the activation of the SPHK1 promoter in hepatoma cells. AP2α siRNA could decrease the expression of SPHK1 induced by HBx in HepG2-X cells. Moreover, the ChIP assay demonstrated that AP2α could bind to the AP2α element in the promoter of SPHK1.
It has been reported that HBx plays a crucial role in transactivation, involving many important oncogenes, transcription factors and signal pathways, such as PTEN, P53, YAP, NF-κB, PI3K/mTOR, in hepatocarcinogenesis9,38,39,40,41,42. Therefore, we assessed whether HBx can transactivate AP2α to activate the promoter of SPHK1. Accordingly, hepatitis B virus regulates Raf1 promoter activity through activation of transcription factor AP2α26, and we validated that HBx was able to upregulate AP2α in hepatoma cells, which is consistent with this report. Thus, our data suggest that HBx enhances the expression of SPHK1 by upregulation of AP2α in hepatoma cells. Functionally, we demonstrated that SPHK1 siRNA remarkably inhibited the growth of hepatoma mediated by HBx in vitro and in vivo. Our data are consistent with the reports that SPHK1 is able to promote tumor cell proliferation17,43. Therefore, HBx modulates SPHK1 via upregulating transcriptional factor AP2α in promotion of hepatoma cell growth. Therapeutically, SPHK1 may serve as a target in liver cancer.
Author contribution
Xiao-dong ZHANG, Zhan-ping LU, and Li-hong YE designed the research; Zhan-ping LU, Ze-lin XIAO, Zhe YANG, Jiong LI, Guo-xing FENG, Fu-quan CHEN, Ying-hui LI, Jin-yan FENG, and Yu-en GAO performed the research; Zhan-ping LU and Ze-lin XIAO analyzed the data; Xiao-dong ZHANG and Zhan-ping LU wrote the manuscript.
References
Buendia MA, Neuveut C . Hepatocellular carcinoma. Cold Spring Harb Perspect Med 2015; 5.
Zhang XD, Wang Y, Ye LH . Hepatitis B virus X protein accelerates the development of hepatoma. Cancer Biol Med 2014; 11: 182–90.
Kong GY, Zhang JP, Zhang S, Shan CL, Ye LH, Zhang XD . Hepatitis B virus X protein promotes hepatoma cell proliferation via upregulation of MEKK2. Acta Pharmacol Sin 2011; 32: 1173–80.
Xie Q, Chen L, Shan X, Shan X, Tang J, Zhou F, et al. Epigenetic silencing of SFRP1 and SFRP5 by hepatitis B virus X protein enhances hepatoma cell tumorigenicity through Wnt signaling pathway. Int J Cancer 2014; 135: 635–46.
Kim HY, Jung HU, Yoo SH, Yoo KS, Cheong J, Park BS, et al. Sorafenib overcomes the chemoresistance in HBx-expressing hepatocellular carcinoma cells through down-regulation of HBx protein stability and suppresses HBV gene expression. Cancer Lett 2014; 355: 61–9.
Martin-Lluesma S, Schaeffer C, Robert EI, van Breugel PC, Leupin O, Hantz O, et al. Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 2008; 48: 1467–76.
You X, Liu F, Zhang T, Li Y, Ye L, Zhang X . Hepatitis B virus X protein upregulates oncogene Rab18 to result in the dysregulation of lipogenesis and proliferation of hepatoma cells. Carcinogenesis 2013; 34: 1644–52.
You X, Liu F, Zhang T, Lv N, Liu Q, Shan C, et al. Hepatitis B virus X protein upregulates Lin28A/Lin28B through Sp-1/c-Myc to enhance the proliferation of hepatoma cells. Oncogene 2014; 33: 449–60.
Zhang T, Zhang J, You X, Liu Q, Du Y, Gao Y, et al. Hepatitis B virus X protein modulates oncogene Yes-associated protein by CREB to promote growth of hepatoma cells. Hepatology 2012; 56: 2051–9.
Du Y, Kong G, You X, Zhang S, Zhang T, Gao Y, et al. Elevation of highly up-regulated in liver cancer (HULC) by hepatitis B virus X protein promotes hepatoma cell proliferation via down-regulating p18. J Biol Chem 2012; 287: 26302–11.
Zhang T, Zhang J, Cui M, Liu F, You X, Du Y, et al. Hepatitis B virus X protein inhibits tumor suppressor miR-205 through inducing hypermethylation of miR-205 promoter to enhance carcinogenesis. Neoplasia 2013; 15: 1282–91.
Shan C, Xu F, Zhang S, You J, You X, Qiu L, et al. Hepatitis B virus X protein promotes liver cell proliferation via a positive cascade loop involving arachidonic acid metabolism and p-ERK1/2. Cell Res 2010; 20: 563–75.
Zhang CX, He HW, Shao RG . Sphingosine kinase 1 and tumor. Yao Xue Xue Bao 2013; 48: 971–8.
Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013; 23: 107–20.
Rosa R, Marciano R, Malapelle U, Formisano L, Nappi L, D'Amato C, et al. Sphingosine kinase 1 overexpression contributes to cetuximab resistance in human colorectal cancer models. Clin Cancer Res 2013; 19: 138–47.
Bao M, Chen Z, Xu Y, Zhao Y, Zha R, Huang S, et al. Sphingosine kinase 1 promotes tumour cell migration and invasion via the S1P/EDG1 axis in hepatocellular carcinoma. Liver Int 2012; 32: 331–8.
Datta A, Loo SY, Huang B, Wong L, Tan SS, Tan TZ, et al. SPHK1 regulates proliferation and survival responses in triple-negative breast cancer. Oncotarget 2014; 5: 5920–33.
Zhang H, Wang Q, Zhao Q, Di W . MiR-124 inhibits the migration and invasion of ovarian cancer cells by targeting SphK1. J Ovarian Res 2013; 6: 84.
Zhang Z, Yan Z, Yuan Z, Sun Y, He H, Mai C . SPHK1 inhibitor suppresses cell proliferation and invasion associated with the inhibition of NF-kappaB pathway in hepatocellular carcinoma. Tumour Biol 2015; 36: 1503–9.
Kapitonov D, Allegood JC, Mitchell C, Hait NC, Almenara JA, Adams JK, et al. Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res 2009; 69: 6915–23.
Wang Y, Cui F, Lv Y, Li C, Xu X, Deng C, et al. HBsAg and HBx knocked into the p21 locus causes hepatocellular carcinoma in mice. Hepatology 2004; 39: 318–24.
Zhang W, Lu Z, Kong G, Gao Y, Wang T, Wang Q, et al. Hepatitis B virus X protein accelerates hepatocarcinogenesis with partner survivin through modulating miR-520b and HBXIP. Mol Cancer 2014; 13: 128.
Shan C, Zhang S, Cui W, You X, Kong G, Du Y, et al. Hepatitis B virus X protein activates CD59 involving DNA binding and let-7i in protection of hepatoma and hepatic cells from complement attack. Carcinogenesis 2011; 32: 1190–7.
Liu Y, Saiyan S, Men TY, Gao HY, Wen C, Liu Y, et al. Hepatopoietin Cn reduces ethanol-induced hepatoxicity via sphingosine kinase 1 and sphingosine 1-phosphate receptors. J Pathol 2013; 230: 365–76.
Cui M, Xiao Z, Wang Y, Zheng M, Song T, Cai X, et al. Long noncoding RNA HULC modulates abnormal lipid metabolism in hepatoma cells through an miR-9-mediated RXRA signaling pathway. Cancer Res 2015; 75: 846–57.
Qu J, Li J, Chen K, Qin D, Li K, Sheng Y, et al. Hepatitis B virus regulation of Raf1 promoter activity through activation of transcription factor AP-2alpha. Arch Virol 2013; 158: 887–94.
Zhang X, Ye LH, Zhang XD . A mutant of hepatitis B virus X protein (HBx Delta 127) enhances hepatoma cell migration via osteopontin involving 5-lipoxygenase. Acta Pharmacol Sin 2010; 31: 593–600.
Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010; 465: 1084–8.
Puneet P, Yap CT, Wong L, Lam Y, Koh DR, Moochhala S, et al. SphK1 regulates proinflammatory responses associated with endotoxin and polymicrobial sepsis. Science 2010; 328: 1290–4.
Xu M, Liu D, Ding LH, Ma KL, Wu M, Lv LL, et al. FTY720 inhibits tubulointerstitial inflammation in albumin overload-induced nephropathy of rats via the Sphk1 pathway. Acta Pharmacol Sin 2014; 35: 1537–45.
Yang L, Chang N, Liu X, Han Z, Zhu T, Li C, et al. Bone marrow-derived mesenchymal stem cells differentiate to hepatic myofibroblasts by transforming growth factor-beta1 via sphingosine kinase/sphingosine 1-phosphate (S1P)/S1P receptor axis. Am J Pathol 2012; 181: 85–97.
Liu SQ, Su YJ, Qin MB, Mao YB, Huang JA, Tang GD . Sphingosine kinase 1 promotes tumor progression and confers malignancy phenotypes of colon cancer by regulating the focal adhesion kinase pathway and adhesion molecules. Int J Oncol 2013; 42: 617–26.
Schulte JH, Kirfel J, Lim S, Schramm A, Friedrichs N, Deubzer HE, et al. Transcription factor AP2alpha (TFAP2α) regulates differentiation and proliferation of neuroblastoma cells. Cancer Lett 2008; 271: 56–63.
Shi D, Xie F, Zhang Y, Tian Y, Chen W, Fu L, et al. TFAP2A regulates nasopharyngeal carcinoma growth and survival by targeting HIF-1alpha signaling pathway. Cancer Prev Res (Phila) 2014; 7: 266–77.
Murakami M, Ichihara M, Sobue S, Kikuchi R, Ito H, Kimura A, et al. RET signaling-induced SPHK1 gene expression plays a role in both GDNF-induced differentiation and MEN2-type oncogenesis. J Neurochem 2007; 102: 1585–94.
Ding X, Yang Z, Zhou F, Wang F, Li X, Chen C, et al. Transcription factor AP-2alpha regulates acute myeloid leukemia cell proliferation by influencing Hoxa gene expression. Int J Biochem Cell Biol 2013; 45: 1647–56.
Chen C, Guo L, Shi M, Hu M, Hu M, Yu M, et al. Modulation of IFN-gamma receptor 1 expression by AP-2alpha influences IFN-gamma sensitivity of cancer cells. Am J Pathol 2012; 180: 661–71.
Knoll S, Furst K, Thomas S, Villanueva Baselga S, Stoll A, Schaefer S, et al. Dissection of cell context-dependent interactions between HBx and p53 family members in regulation of apoptosis: a role for HBV-induced HCC. Cell Cycle 2011; 10: 3554–65.
Yang ST, Yen CJ, Lai CH, Lin YJ, Chang KC, Lee JC, et al. SUMOylated CPAP is required for IKK-mediated NF-kappaB activation and enhances HBx-induced NF-kappaB signaling in HCC. J Hepatol 2013; 58: 1157–64.
Ortiz-Cuaran S, Villar S, Gouas D, Ferro G, Plymoth A, Khuhaprema T, et al. Association between HBX status, aflatoxin-induced R249S TP53 mutation and risk of hepatocellular carcinoma in a case-control study from Thailand. Cancer Lett 2013; 331: 46–51.
Chung TW, Lee YC, Ko JH, Kim CH . Hepatitis B virus X protein modulates the expression of PTEN by inhibiting the function of p53, a transcriptional activator in liver cells. Cancer Res 2003; 63: 3453–8.
Zhu M, Guo J, Li W, Lu Y, Fu S, Xie X, et al. Hepatitis B virus X protein induces expression of alpha-fetoprotein and activates PI3K/mTOR signaling pathway in liver cells. Oncotarget 2015; Jan 21 [Epub ahead of print].
Kalari S, Moolky N, Pendyala S, Berdyshev EV, Rolle C, Kanteti R, et al. Sphingosine kinase 1 is required for mesothelioma cell proliferation: role of histone acetylation. PLoS One 2012; 7: e45330.
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (973 Program, No 2015CB553703, 2015CB553905), the National Natural Science Foundation of China (No 81272218, 81372186) and the Project of Prevention and Treatment of Key Infectious Diseases (No 2014ZX0002002-005).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary tables and figures are available at the Acta Pharmacologica Sinica website.
Rights and permissions
About this article

Cite this article
Lu, Zp., Xiao, Zl., Yang, Z. et al. Hepatitis B virus X protein promotes human hepatoma cell growth via upregulation of transcription factor AP2α and sphingosine kinase 1. Acta Pharmacol Sin 36, 1228–1236 (2015). https://doi.org/10.1038/aps.2015.38
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2015.38
Keywords
This article is cited by
-
Integrative roles of sphingosine kinase in liver pathophysiology
Toxicological Research (2023)
-
miR-511 promotes the proliferation of human hepatoma cells by targeting the 3′UTR of B cell translocation gene 1 (BTG1) mRNA
Acta Pharmacologica Sinica (2017)
