
Abstract
Autophagy, an evolutionarily conserved catabolic process involving the engulfment and degradation of non-essential or abnormal cellular organelles and proteins, is crucial for homeostatic maintenance in living cells. This highly regulated, multi-step process has been implicated in diverse diseases including cancer. Autophagy can function as either a promoter or a suppressor of cancer, which makes it a promising and challenging therapeutic target. Herein, we overview the regulatory mechanisms and dual roles of autophagy in cancer. We also describe some of the representative agents that exert their anticancer effects by regulating autophagy. Additionally, some emerging strategies aimed at modulating autophagy are discussed as having the potential for future anticancer drug discovery. In summary, these findings will provide valuable information to better utilize autophagy in the future development of anticancer therapeutics that meet clinical requirements.
Similar content being viewed by others


Introduction
Macroautophagy (hereafter referred to as “autophagy”) is an evolutionarily conserved and dynamically catabolic process in which a cell degrades misfolded and compromised proteins and damaged organelles1. It was first proposed to be an important cellular process by Deter and De Duve almost 55 years ago2 and was classified by Clarke as an alternative, non-apoptotic route of programmed cell death (PCD) referred to as “type II PCD” in 19903. The morphological changes observed in the initial stages of the autophagic process involve the appearance of intracellular double-membrane autophagic vacuoles, called autophagosomes4. These autophagosomes then mature by fusing with lysosomes to form autolysosomes5 in which the sequestered bulk cytoplasm, long-lived proteins and cellular organelles are degraded by lysosomal hydrolases, thus leading to nutrient recycling and increased cellular energy.
Under normal conditions, autophagy remains at a low basal level, which is essential for the maintenance of cellular homeostasis via the turnover of energy and cellular building material1,6. However, dysregulated autophagy could be implicated in diverse pathological processes, such as metabolic diseases7, neurodegenerative diseases8,9, infectious diseases10, inflammatory diseases11, aging12,13 and cancer14. Although the potential of autophagy as a therapeutic target for some diseases has been validated by numerous investigations15,16, the utilization of autophagy in anticancer treatment appears to be more complex due to its dual role in response to specific cancer microenvironments17,18. On the one hand, as a tumor suppressor, autophagy could negatively regulate tumorigenesis and progression, and thus the excessive and sustained induction of autophagy may limit the survival of cancer cells. On the other hand, given the tumor-promoting activity of autophagy in cancer, the inhibition of cytoprotective autophagy could also exert anticancer effects under certain contexts. In addition, the intrinsic crosstalk between autophagy and apoptosis gives rise to additional opportunities to improve anticancer therapeutics19. Accumulating data has demonstrated the efficiency of existing autophagy modulating agents in anticancer treatment, and the emergence of some other therapeutic strategies may launch a new wave of drug discovery through the targeting of autophagy.
In this review, we briefly describe the regulation of the autophagy process, discuss the double-edged sword of autophagy in cancer, summarize current autophagy-modulating agents for anticancer treatment and put forward emerging strategies for further anticancer drug discovery by targeting autophagy, in the hope of better utilizing autophagy in future anticancer therapeutics.
A brief overview of autophagy in cancer
The regulation of autophagy in cancer
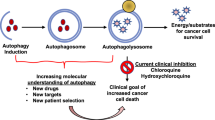
Autophagy is readily induced in response to nutrient deprivation, metabolic stress, endoplasmic reticulum (ER)-stress, radiation and anticancer drugs. In general, the complete autophagic process can be divided into several stages, including induction, vesicle nucleation, vesicle elongation and completion, docking and fusion, degradation and recycling20 (Figure 1). Although the molecular basis of autophagy was discovered in 199321, the intricate link between autophagy and cancer was not established until 1999, when the autophagy gene Beclin1 (Becn1; also known as Atg6) was found to inhibit tumorigenesis and was assumed to be a candidate tumor suppressor22. Then it followed that the genomic deletion of Becn1 or the downregulation of Beclin1 might be able to contribute to the initiation or the development of certain cancers. Additionally, other autophagy-related proteins, such as Bcl-2, vacuolar sorting protein 34 (Vps34), ultraviolet irradiation resistance-associated gene (UVRAG), Bif1 and Atg14L, can bind with Beclin 1 to form Beclin 1 interactome, and further promote the initiation of autophagy23.

The process of autophagy. Under conditions of nutrient deprivation, metabolic stress, ER stress, radiation or anticancer drug treatment, autophagy is readily induced. The complete autophagic flow can be divided into several stages: induction, vesicle nucleation, vesicle elongation and completion, docking and fusion, degradation and recycling.
In most cases, the induction of autophagy is closely related to the mammalian rapamycin complex 1 (mTORC1), a central controller of cell growth24. Under certain circumstances, the suppression of mTORC1 can trigger autophagic cascades to aid in the survival of metabolic or hypoxic stress. However, the activation of mTORC1 can negatively regulate autophagy by phosphorylating a complex of autophagy proteins such as the Unc51-like kinases (ULK1/2), which interfere with the formation of autophagosomes25. AMP-activated protein kinase (AMPK), a central sensor of cellular nutrient status or energy levels, is one of the upstream regulators of mTORC1. Nutrient deprivation leads to the activation of AMPK, which then activates tuberous sclerosis protein 2 (TSC2) to repress mTORC1 and upregulate autophagy. The phosphatidyl inositol-3-kinase (PI3K)-Akt pathway, which is frequently dysregulated in human cancers, is another important pathway that signals upstream of mTORC1. This pathway can downregulate the expression of the TSC1/TSC2 complex, a tumor suppressor complex existing in various cancer types. The PI3K-Akt pathway further suppresses mTORC1 by inactivating the mTORC1-interacting protein Rheb (Ras homolog enriched in brain), thereby playing a pro-survival role in cancer cells26,27. Moreover, the PI3K-Akt-mTORC1 axis can also be regulated indirectly by the activation of Ras, which interacts with the p110α catalytic subunit of PI3K and strengthens the effects of the Ras-Raf-MAPK (mitogen-activated protein kinase) pathway in cancer28. Activated Ras binds to Raf and subsequently phosphorylates mitogen-activated protein kinase 1/2 (MEK1/2) and extracellular signal-regulated kinase 1/2 (ERK1/2).
In addition, roles for the well-known tumor suppressor gene TP53 in the regulation of autophagy are paradoxical, depending on the subcellular localization of the p53 protein. Nuclear p53 functions mainly by triggering the transcription of several autophagy inducers, such as damage-regulated autophagy modulator (DRAM)29, Sestrin230, Bcl-2-associated X protein (Bax) and p53-upregulated modulator of apoptosis (PUMA)31, thus positively regulating autophagy. Moreover, the activation of autophagy by nuclear p53 may also be related to AMPK- and TSC1/TSC2-dependent mTORC1 inhibition32. However, genetic or pharmacological loss of p53 function can also activate autophagy, suggesting the negative regulation of autophagy by cytoplasmic p5333. To date, the dual interplay between p53 and autophagy remains unclear, making it challenging to target p53 for the modulation of autophagy.
Aside from the above-mentioned classical regulators, other mechanisms such as ER stress can also induce autophagic cell death34, indicating that it is worthwhile to exploit this adaptive mechanism for the benefit of cancer treatment. ER stress is often accompanied by the release of calcium into the cytosol; during this process, calcium- and calmodulin-dependent protein kinase kinase (CAMKK) β-dependent AMPK activation can further connect calcium release from the ER to autophagy35. Some major autophagic regulators and related pathways that play crucial roles in the regulation of autophagy in cancer, including Beclin 1 interactome, the PI3K-Akt-mTOR pathways, the Ras-Raf-MAPK pathways and TP53 signaling are shown in Figure 2. Taken together, multiple molecules and signaling pathways could modulate autophagy in cancer, and these regulators may serve as potential therapeutic targets in cancer.

Core signaling pathways regulating autophagy in cancer. Some major autophagic regulators and related pathways, including Beclin 1 interactome, p53 signaling, PI3K-Akt-mTOR pathways, and Ras-Raf-MEK-ERK pathways, play crucial roles in the regulation of autophagy in cancer.
Crosstalk between autophagy and apoptosis in cancer
Apoptosis [a term from Greek “apo” (from) and “ptosis” (falling)], or type I PCD, is an evolutionarily conserved mechanism of cell death that may occur in response to various physiological and pathological events. This biochemical event leads to morphological changes in dying cells including cell shrinkage, nuclear DNA fragmentation, membrane blebbing and eventually the formation of apoptotic bodies36. Although autophagy and apoptosis have distinct morphological and biochemical characteristics, they still share some common regulatory factors and components and exert overlapping physiological functions, leading to intricate interactions between them.
Recently, studies have indicated that some important regulators, such as p53, the PI3K/Akt axis and the interaction between Bcl-2 and Beclin-1, could synchronously control apoptosis and autophagy37. Under certain conditions, upstream signals could result in the simultaneous activation of autophagy and apoptosis38. Moreover, some positive or negative feedback between autophagy and apoptosis exists. For example, the activation of autophagy could either inhibit apoptosis39 or contribute to its induction40. Of note, apoptosis is often inactivated in tumor cells, which facilitates tumor progression and contributes to tumor cell resistance to radiotherapy and chemotherapy. Therefore, an activation of autophagy when apoptosis is blocked could function as a primary form of cell death in apoptosis-deficient cells41. Accordingly, while the mechanisms underlying the intrinsic link between autophagy and apoptosis require further investigation, their crosstalk could undoubtedly provide more opportunities for autophagy-targeted anticancer therapeutics.
The double-edged sword of autophagy in cancer
Although the pivotal role of autophagy in cancer is indisputable, considering the complexity of tumor microenvironment and autophagy regulation, the relationships between autophagy and cancer appear to vary, depending on specific cellular and extra-cellular context42. During the early stages of tumorigenesis, autophagy acts as a tumor suppressor by preventing tumor initiation. Paradoxically, in established tumors, autophagy acts as a tumor promoter by facilitating their survival under stressful conditions induced by nutrient starvation or chemotherapeutic agents. Thus, a better understanding of the underlying dual mechanisms of autophagy in tumor initiation and progression is essential to better harness autophagy in cancer treatment.
Autophagy and tumor suppression
The tumor suppressive role for autophagy was first documented by the discovery that Beclin 1, an upstream regulator of the autophagic pathway, is a potential tumor suppressor in breast cancer22. Recent studies have shown that the overexpression of this gene can inhibit the growth of colon cancer cells43 and CaSki cervical cancer cells44, whereas the downregulation of this gene has been observed in lung cancer45, esophageal cancer46 and gastric cancer47. Moreover, genes encoding other components of autophagic pathways, including Atg2B, Atg5, Atg9B, and Atg12, have also been demonstrated to undergo chromosomal deletions or show decreased expression in specific cancer types48, suggesting suppressive roles for these core autophagy genes in tumorigenesis.
Although the definite mechanism by which autophagy acts as a tumor suppressor still remains unclear, some hypotheses have been put forth to explain the roles of autophagy in tumor suppression. First, autophagy can prevent organelle normality, chromosomal stability and genomic integrity at early stages of tumorigenesis. A recent study reported that enhanced autophagy induced by the upregulation of microtubule-associated protein 1S (MAP1S) could suppress DNA double-strand breaks and genome instabilities and thus repress the incidence of hepatocarcinogenesis49. Second, autophagy may facilitate oncogene-induced senescence (OIS)50, which is a major tumorigenesis barrier that causes permanent cell cycle arrest and inhibits the proliferation of cancer cells. Third, autophagy could serve as an action mechanism for radiation therapy and chemotherapy. Some studies have shown that the induction of autophagy contributes to radiation sensitization in both malignant glioma cells and radio-resistant glioma cells51,52. Studies have also demonstrated that some chemotherapeutic drugs might kill tumor cells via the intrinsic apoptosis pathway induced by autophagy53.
Autophagy and tumor progression
Remarkably, elevated autophagy has been observed in many cancer cells under basal conditions54, suggesting that autophagy might be essential for tumor cell survival during the advanced stages of tumorigenesis. Several mechanisms have been put forward to explain the tumor-promoting role of autophagy. First, autophagy may be essential for cancer cells to maintain genomic stability and organelle homeostasis. A previous study has shown that the inhibition of autophagy can lead to increased reactive oxygen species (ROS), elevated DNA damage and a metabolic defect in pancreatic cancer cell lines55. Additionally, autophagy-mediated degradation and recycling could provide key intermediates that meet the metabolic requirements for the promotion of tumor survival and progression56. Under nutrient deprivation or other stressful conditions, robustly induced autophagy could result in nutrient recycling and increased energy, thus facilitating cancer cells to survive even under those stressful conditions57. Finally, autophagy may be a crucial resistance mechanism for tumor maintenance in response to cancer therapies such as radiation and chemotherapy.
A recent study disrupted autophagy and observed that the cells rapidly underwent apoptosis, demonstrating that BCR-ABL-mediated leukemogenesis in a hematopoietic cell culture is highly dependent on the basal levels of autophagy58. Another research study has reported that the resistance of some aggressive cancers to chemotherapeutic drugs largely relies on astrocyte-elevated gene-1 (AEG-1), which mediates protective autophagy and thus limits drug efficiency59. These studies illustrate the necessity of autophagy-mediated survival in tumor progression and the potential of anti-autophagy therapies in cancer treatment.
To summarize, autophagy can serve dual functions in cancer by acting as a tumor-suppressing pathway that prevents tumor initiation in the early stage of tumorigenesis and as a tumor-promoting pathway that helps established tumor cells to survive stressful conditions. Together, these make autophagy a challenging but promising target for anticancer therapeutics. When exploring the modulation of autophagy for anticancer therapy, the stages of tumorigenesis, the specific cellular and extra-cellular context and the crosstalk between autophagy and apoptosis should all be taken into account.
Autophagy modulation for anticancer treatment
The seemingly paradoxical tumor-suppressing and -promoting roles of autophagy provide more therapeutic opportunities for anticancer treatment. On the one hand, the features of autophagy are usually observed in dying cancer cells that have been exposed to chemotherapy and radiotherapy, suggesting that autophagy may facilitate or at least accompany cancer cell death60. Importantly, given that inactivated apoptosis is a common mechanism of cancer therapy resistance, the induction of autophagic cell death could be an effective approach to suppress tumor growth and to alleviate treatment resistance in apoptosis-defective tumor cells. On the other hand, high basal levels of autophagy in tumor cells could play a critical role for rapid tumor proliferation by meeting metabolic demands and eliminating unnecessary or dysfunctional cellular components. Furthermore, in response to stresses induced by cancer therapies, an upregulation of autophagy is also likely to contribute to the survival of tumor cells by driving therapy resistance61, thus opening new perspectives for anticancer therapeutics by inhibiting autophagy. Currently, a number of autophagy-modulating agents have been proposed to be potential anticancer therapeutics when used as either single or combinatorial treatments.
Autophagy inducers in cancer therapy
Previously, various agents targeting different signaling pathways or cellular processes in cancer cells have been suggested to induce autophagy. For example, several inhibitors of mTOR62,63,64,65,66, tyrosine kinases67,68,69,70,71, Akt72,73,74 and activators of the cell energy sensor AMPK75,76 can lead to an induction of autophagy in cancer cells. Moreover, agents regulating cellular process such as the ubiquitin-proteasome77,78,79,80, histone deacetylation81,82,83,84,85,86,87, apoptosis88,89,90,91 and other therapies92,93,94,95,96,97 can also trigger autophagy in multiple tumor types as selectively described in Table 1.
Several allosteric and catalytic inhibitors of mTOR could exert their antitumor effects by inducing autophagy. Rapamycin, an allosteric inhibitor of mTORC1, can selectively inhibit mTORC1 by binding to the 12 kDa immunophilin FK506-binding protein (FKBP12) to stimulate autophagy62. Its analogs, temsirolimus (CCI-779), everolimus (RAD001) and deforolimus (AP-23573), can also target mTORC1 for the treatment of cancer while not targeting mTORC263,64,65,66. In addition to stimulating autophagic cancer cell death, the induction of autophagy by mTOR inhibitors could also sensitize cancer cells to other cancer therapeutics.
Additionally, specific tyrosine kinase inhibitors have been suggested to be effective anticancer therapies that act by triggering autophagy. Imatinib, a potent inhibitor of BCR-ABL, KIT, and platelet-derived growth factor (PDGFR), can induce autophagy in chronic myeloid leukemia (CML) cells through the Becn1 and Atg5 genes, thus potentiating imatinib-induced cell death67. Another BCR-ABL inhibitor, INNO-406, can result in autophagic cell death in Bcr-Abl-positive leukemias69. Furthermore, dasatinib, a SRC-ABL inhibitor, can exert its antitumor effects in ovarian cancer by stimulating autophagic cell death70. However, it should be noted that autophagy may also be a survival mechanism underlying resistance to imatinib and dasatinib68,71.
Recently, preclinical studies have shown that Akt inhibitors such as perifosine72 and triciribine73 can induce protective autophagy in tumor cells, and perifosine can enhance the cytotoxicity of rapamycin in multiple myeloma through the suppression of the PI3K/Akt/mTOR pathway74. Moreover, studies have demonstrated that metformin can activate AMPK and thus suppress tumor progression in cervical cancer75 and melanoma76 by triggering autophagy. These findings confirm the efficiency of this metabolism agonist in autophagy-related cancer therapeutics.
The ubiquitin-proteasome system (UPS) and autophagy are two major intracellular protein degradation systems critical for cellular function, and accumulating evidence has confirmed a coordinated and complementary relationship between the two systems77. Proteasome inhibition by bortezomib, which is approved for the treatment of multiple myeloma, can initiate autophagy through the activation of jun N-terminal kinase (JNK) pathways in head and neck squamous cell carcinoma (HNSCC) cells78. Moreover, in HeLa cervical carcinoma cells, bortezomib has been demonstrated to exert its cytotoxic effect by activating autophagy, blocking apoptosis, and by eventually leading to necrosis79. Interestingly, proteasome inhibitor-induced autophagy appears to be a compensatory response to disabled proteasome function. For example, bortezomib could activate autophagy as a cytoprotective mechanism to eliminate protein aggregates and alleviate proteotoxic stress in human prostate cancer cells, and the simultaneous blockage of proteasome and autophagy could result in enhanced cytotoxicity compared to the inhibition of either degradation pathway alone80. This suggests that the combined inhibition of autophagy and proteasome could be an attractive strategy for anticancer therapeutics.
Histone deacetylase (HDAC), a class of enzymes essential to the regulation of DNA transcription through the modulation of histone deacetylation, has been validated to be critical for the management of autophagy. Thus, the stimulation of autophagy by targeting HDAC could be another candidate therapeutic strategy; suberoylanilide hydroxamic acid (SAHA), a well known HDAC inhibitor, can trigger caspase-independent autophagic cell death in endometrial stromal sarcoma cells83. Recently, a new synthetic HDAC inhibitor, MHY218, has been demonstrated to decrease the expression levels of HDAC1, 4, and 6, leading to autophagy-related cell death in tamoxifen-resistant MCF-7 breast cancer cells85. Of note, HDAC6 is necessary for the activation of autophagy when the proteasome system is impaired; thus, the combination of proteasome inhibitors and HDAC inhibitors could be a promising anticancer strategy, which has been validated in the treatment of cervical cancer86 and HNSCC87.
The crosstalk between apoptosis and autophagy could also be exploited to better harness autophagy for anticancer therapeutics. Evidence has shown that some inhibitors of Bcl-2 that target the BH-3 domain, which is essential for the interaction between Beclin1 and Bcl-2/Bcl-xL, could disrupt the interplay between autophagy and apoptosis to induce autophagy88. For example, a novel compound targeting the BH3 binding groove of Bcl-xL/Bcl-2, Z18, could efficiently induce autophagic cell death in HeLa cells without apparent apoptosis89. Additionally, a natural BH3-mimetic, (–)-gossypol, can activate the autophagic pathway and inhibit the growth of apoptosis-resistant prostate cancer xenograft models90. Moreover, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) can induce apoptotic cell death in various tumor types, while in TRAIL-resistant cancer cells, a human antibody against the TRAIL receptor 2, HW1, has been demonstrated to provide a new therapeutic strategy by triggering autophagic cell death91.
Furthermore, under some circumstances, inducers of autophagy could be utilized to potentiate other anticancer therapeutics. For example, although RAD001 alone showed no therapeutic effect for ovarian cancer, when combined with arsenic trioxide (As2O3), a synergistic tumor inhibitory effect along with an induction of autophagy and apoptosis was observed in ovarian cancer xenograft models92. Moreover, RAD001 could augment the cytotoxic effects of radiation in prostate cancer cells64, and the combined use of rapamycin and apoptosis inducers can enhance radiation therapy in nonsmall cell lung cancer (NSCLC) cell lines and xenograft models93. Additionally, the combined treatment of metformin with the mTORC1 inhibitor temsirolimus can suppress the growth of lymphoma cells through the induction of autophagy and act more efficiently than either agent alone94, thus providing additional therapeutic opportunities by exploiting the induction of autophagy for anticancer treatment.
Autophagy inhibitors in cancer therapy
Given the cytoprotective role of autophagy in cancer, therapeutic strategies involving its inhibition have also been demonstrated to have potential based on existing experimental data. Representative autophagy inhibitors developed for the purposes of suppressing tumor development are described in Table 1.
Class-III-PI3K inhibitors [eg, 3-methyladenine (3-MA)] can interfere with the formation of autolysosomes and suppress the migration and invasion of HT1080 fibrosarcoma cells98. Additionally, the inhibition of autophagy by 3-MA could enhance the therapeutic effects of 5-fluorouracil (5-FU) in colon cancer cells99, and the combined use of 3-MA and cisplatin could also strengthen the apoptotic cell death induced by cisplatin in esophageal squamous cell carcinoma cells100. Furthermore, the cytotoxic effects of radiation could be augmented by 3-MA in radio-resistant HepG2 cell lines and xenograft models101, suggesting the potential for 3-MA as an adjuvant therapeutic strategy under radio-resistant conditions.
Compared to PI3K inhibitors, bafilomycin A1, which is a specific inhibitor of vacuolar-ATPase, can inhibit autophagy by blocking the fusion of autophagosomes and lysosomes102. A recent study has demonstrated that the inhibition of autophagy by bafilomycin A1 could induce cell cycle arrest and apoptosis in colon cancer cells103. Moreover, the inhibition of autophagy by bafilomycin A1 could enhance the cytoxicity of other anticancer therapeutics. For example, treatment with bafilomycin A1 (but not 3-MA) and sulforaphane could significantly strengthen sulforaphane-induced apoptosis in breast cancer cells104; therefore, the combined use of bafilomycin A1 and conventional chemo/radiotherapy could be a potential anticancer strategy.
The widely used anti-malarial and anti-rheumatic drugs chloroquine (CQ) and its analog hydroxychloroquine (HCQ) can block lysosomal acidification and inhibit the degradation of autophagosomes, thereby blocking the late stage of autophagy. A previous study demonstrated that CQ-mediated autophagic cell death may be p53-dependent and is at least in part due to the intrinsic apoptotic pathway in Myc-driven lymphoma105, whereas in glioma cells, the autophagic vacuole accumulation and cell death induced by CQ appears to be p53-independent106. Moreover, when combining CQ with NVP-BEZ235, a dual PI3K/mTOR inhibitor, enhanced anticancer effects induced by the interruption of autophagy were observed in glioblastoma cells and non-small cell lung cancer xenograft models107,108. Furthermore, CQ or HCQ has also been suggested to strengthen the efficiency of certain chemotherapeutic drugs. For example, in colon cancer cells, the anticancer effects of 5-FU were augmented by combined treatment with CQ109. However, a recent study has reported that the enhancement of chemotherapy efficiency by CQ might be independent of autophagy110, which warrants caution for future investigations targeting the roles of CQ in combined cancer therapy111. Currently, many phase I/II clinical trials are ongoing to evaluate CQ- and HCQ-mediated modulation of autophagy in combination with standard treatment in patients with breast, colorectal, hematologic, kidney, lung, ovarian, pancreatic and prostate cancers (Table 2). If these trials achieve satisfactory results, these therapeutic strategies will undoubtedly bring renewed interest to the present anticancer therapeutic market.
Taken together, the studies described above suggest that various agents can target different events involved in the activation or inhibition of autophagy and ultimately culminate in tumor suppression. However, although several existing therapeutic strategies for modulating autophagy have proven to be effective in cell culture systems, much work remains to be done to determine the specific impact of these modulators on tumor initiation, promotion, progression and therapeutic response for further clinical uses. Thus, the acceleration of drug discovery and development to exploit more selective agents that can efficiently modulate autophagy either alone or in combination with other treatments is imperative for better utilization of autophagy for anticancer therapeutics.
Emerging strategies in autophagy-targeted anticancer drug discovery
Given the complexity of autophagy-driving mechanisms and the dual roles for autophagy in cancer, the molecular pharmacology of autophagy is faced with challenges. Nowadays, several reasonable approaches to drug discovery are emerging to allow for a deeper exploration of the therapeutic potentials of autophagy in cancer. Here, we describe some potential strategies, such as systems biology approaches, microRNA (miRNA) modulation and microtubule-associated protein 1A/1B-light chain 3 (LC3)-based screening assays, that may be informative for the discovery of new drugs in autophagy-related anticancer therapeutics.
Systems biology approaches for the discovery of novel regulators of autophagy
From the systems biology perspective, the integration of diverse information from genomics, transcriptomics, proteomics and interactomics may provide essential information to elucidate the mechanisms underlying the regulation of autophagy and to discover novel autophagy regulators in the context of cancer112. A genomics approach combined with a functional RNA interference (RNAi) screen might be useful in capturing novel autophagy modulators. For example, using a genome-wide RNAi screen, researchers have successfully identified 9 novel regulators of starvation-induced autophagy113 and 10 novel autophagy-related kinase candidates in MCF-7 human breast carcinoma cells114. In addition, the integration of proteomic and genetic data sets revealed EEF1G to be a novel regulator of autophagy in both yeast cells and human breast cancer cells115. A proteomics approach using quantitative mass spectrometry could also be effective in identifying novel autophagosomal markers116. Moreover, miRNA microarray profiling may have the potential to screen for miRNAs involved in the control of autophagy. For example, the use of global miRNA expression profiling has demonstrated that miR-30a can negatively regulate autophagy by targeting Beclin-1 in MCF-7 cells117, whereas miR-20a and miR-106b can inhibit autophagy by suppressing ULK1 under physiological conditions118. Interestingly, the combination of miRNA expression profiling and transcriptome profiling also led to the successful identification of autophagy-associated miRNAs and their specific targets119.
Furthermore, biological networks such as protein-protein interaction (PPI) networks may help to illustrate the structures and dynamics of cellular processes rather than the characteristics of the individual parts. Recently, a proteomic network of the human autophagy system under basal autophagy conditions (a network of 751 interactions among 409 candidate interacting proteins) was established120 that provides a global view of the mammalian autophagy interaction landscape and favors further mechanistic analysis of this essential homeostatic process. Network analysis may also be an ideal way to understand autophagy-related cancer networks and to identify candidate targets for the control of autophagy. For example, the construction of a hybrid “yeast-human” core autophagy network led to the identification of 14 novel autophagy-associated proteins121, demonstrating the utility of the network approach to discover novel modulators of autophagy.
MiRNA modulation in autophagy-targeted anticancer therapeutics
MiRNAs are small, non-coding RNAs of ∼22 nucleotides (nt) in length that can inhibit gene expression by blocking the translation or by inducing the degradation of target miRNA122. Recently, accumulating evidence has suggested that miRNAs play critical roles in cell survival/death and therefore show great potential in regulating autophagy at different stages123. During the early stage of autophagy, miR-376b can decrease the activity of Beclin 1, thereby blocking vesicle nucleation124. For autophagosome formation, miR-101 can inhibit Ras-related protein Rab-5A (RAB5A), a GTPase regulating early endocytosis, and further block the autophagy process119. In addition, the vesicle elongation and completion phase has been shown to be negatively regulated by miR-101 and miR-376b via the downregulation of Atg4C and Atg4D, respectively119,124.
Of note, the roles of miRNAs in the regulation of autophagy may have profound implications for cancer therapy. A recent study has reported that miRNA-130a can target ATG2B and DICER1 and thus inhibit autophagy and trigger the death of chronic lymphocytic leukemia cells125. Similarly, the downregulation of Beclin 1 by miR-30a may inhibit autophagy and exert significant impacts on cancer development, progression and treatment117. Interestingly, miR-30a-mediated autophagy has also shown therapeutic implications for imatinib resistance in patients with chronic myelogenous leukemia (CML)126, implying the potential for miRNA-targeted autophagy modulation in cancer therapy. Given the dual roles of autophagy in cancer, miRNAs that regulate corresponding autophagic mediators may be either oncogenic or tumor suppressive123, which enhances the possibilities of the utilization of the miRNA system in cancer biology and pathology by targeting autophagic pathways.
LC3-based screening assays for the identification of autophagy-modulating small molecules
High content screening (HCS) and high throughput screening (HTS), which allow for the rapid phenotypic screen of thousands of chemicals in a variety of in vitro assays, have become powerful tools for identifying small molecule modulators of autophagy127. Most screening assays for autophagy modulators rely on LC3, a well-known autophagy marker that tightly binds to autophagosomal membranes, as an indicator of autophagic activity. When conjugated with green fluorescent protein (GFP), the detection of the fluorescence intensity of GFP-LC3 can be used to quantify autophagosome formation in cells treated with small molecule libraries. For example, based on microscopic quantification of GFP-LC3 fluorescence, a recent HCS study has successfully identified three Food and Drug Administration (FDA)-approved drugs and one pharmacological reagent capable of rapidly inducing autophagy from a library of 3500 chemicals128. However, it should be noted that the accumulation of GFP-LC3 might be caused by either upstream inducers or downstream inhibitors of autophagosome formation. Thus, a double-tagged LC3 such as mCherry-GFP-LC3, which could exhibit both GFP and mCherry signals in autophagosomes but only a mCherry signal in autolysosomes, could be utilized to differentiate between autophagosomes before and after fusion with lysosomes129. By employing GFP-LC3 in the initial screening and mCherry-GFP-LC3 for the secondary screening, Hundeshagen et al identified 38 potential activators and 36 inhibitors of autophagy from 1,120 FDA-approved drugs130. This study is the first report of the use of mCherry-GFP-LC3 in a cell based screen for autophagy modulators.
Moreover, combining GFP-LC3 with other assays could help to distinguish compounds that can truly induce autophagy from those that elevate LC3-GFP by causing cellular damage or by blocking lysosomal functions. For example, in an image-based HPS study, researchers used GFP-LC3 and GFP-Rab7 (a marker of endolysosomal activity) to ensure the influence of potential modulators of autophagy on autophagic flux rather than on the endolysosomal pathway130. In another study, the combination of GFP-LC3 detection with an assay for long-lived protein degradation led to the identification of eight compounds that could induce autophagy and promote long-lived protein degradation from a library of 480 compounds131.
In addition to GFP-LC3-based screening methods, the luciferase (Luc)-based screening assay132 and a phospholipase A2(PLA2)-based screening strategy133, which take advantage of a Luc-LC3 reporter and LC3-PLA2 reporter respectively, have also proven to have the potential to identify novel modulators of autophagy. With the development of techniques and tools to efficiently monitor autophagy, HTS/HCS assays will undoubtedly be helpful in developing new drugs for autophagy-related anticancer therapeutics.
In summary, as viewed from a systems biology perspective, agents targeting the core autophagy modulators and the autophagic networks in cancer cells could be further screened as novel candidate anti-cancer drugs. Furthermore, the additional elucidation of the miRNA-modulated autophagic signaling networks and better use of autophagy marker-based HCS and HTS assays may provide alternative strategies and open new avenues for the current and future development of autophagy-based anticancer therapeutics.
Concluding remarks
Autophagy, a basic catabolic mechanism involving the cellular degradation of unnecessary or dysfunctional cellular components, has been suggested to be a potential therapeutic target for diverse diseases including cancer. The complex, dual nature of autophagy, as either a promoter or inhibitor of tumor formation and progression, leads to opposite therapeutic strategies targeting autophagy induction or inhibition. To date, encouraging progress has been made in the area of autophagy-based anticancer therapeutics, and some autophagy-modulating drugs such as rapamycin and tamoxifen have been approved for clinical use in cancer management; thus, autophagy has been established as a promising therapeutic target in cancer. More importantly, attractive strategies for the identification of autophagy-modulating agents are emerging, which may undoubtedly provide new clues for launching a new wave of anticancer drug discovery. In summary, with a better understanding of the potential molecular mechanisms implicated in autophagy and the intrinsic relationships between autophagy and cancer at the level of the entire autophagy network, the ultimate goal of conquering cancer with autophagy modulation as an adjuvant therapeutic strategy will be achieved in the near future.
References
Yang Z, Klionsky DJ . Eaten alive: a history of macroautophagy. Nat Cell Biol 2010; 12: 814–22.
Deter RL, De Duve C . Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol 1967; 33: 437–49.
Clarke PG . Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990; 181: 195–213.
Tanida I . Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal 2011; 14: 2201–14.
Jegga AG, Schneider L, Ouyang X, Zhang J . Systems biology of the autophagy-lysosomal pathway. Autophagy 2011; 7: 477–89.
Rabinowitz JD, White E . Autophagy and metabolism. Science 2010; 330: 1344–8.
He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 2012; 481: 511–5.
Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008; 4: 295–305.
Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J Neurosci 2009; 29: 13578–88.
Munz C . Macroautophagy during innate immune activation. Front Microbiol 2011; 2: 72.
Levine B, Mizushima N, Virgin HW . Autophagy in immunity and inflammation. Nature 2011; 469: 323–35.
Yamaguchi O, Otsu K . Role of autophagy in aging. J Cardiovasc Pharmacol 2012; 60: 242–7.
Rubinsztein DC, Marino G, Kroemer G . Autophagy and aging. Cell 2011; 146: 682–95.
Choi KS . Autophagy and cancer. Exp Mol Med 2012; 44: 109–20.
Rubinsztein DC, Codogno P, Levine B . Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11: 709–30.
Sridhar S, Botbol Y, Macian F, Cuervo AM . Autophagy and disease: always two sides to a problem. J Pathol 2012; 226: 255–73.
Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17: 654–66.
Yang ZJ, Chee CE, Huang S, Sinicrope FA . The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10: 1533–41.
Liu JJ, Lin M, Yu JY, Liu B, Bao JK . Targeting apoptotic and autophagic pathways for cancer therapeutics. Cancer Lett 2011; 300: 105–14.
Liu B, Cheng Y, Liu Q, Bao JK, Yang JM . Autophagic pathways as new targets for cancer drug development. Chin J Cancer 2010; 31: 1154–64.
Tsukada M, Ohsumi Y . Isolation and characterization of autophagy–defective mutants of Saccharomyces cerevisiae. FEBS Lett 1993; 333: 169–74.
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402: 672–6.
Kang R, Zeh HJ, Lotze MT, Tang D . The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18: 571–80.
Jung CH, Ro SH, Cao J, Otto NM, Kim DH . mTOR regulation of autophagy. FEBS Lett 2010; 584: 1287–95.
Chan EY, Longatti A, McKnight NC, Tooze SA . Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol 2009; 29: 157–71.
Qin L, Wang Z, Tao L, Wang Y . ER stress negatively regulates AKT/TSC/mTOR pathway to enhance autophagy. Autophagy 2010; 6: 239–47.
Yu J, Henske EP . TORC1 activation and autophagy inhibition: opposing forces in Rheb-driven tumorigenesis. Cell Cycle 2011; 10: 3803–4.
Wang SY, Yu QJ, Zhang RD, Liu B . Core signaling pathways of survival/death in autophagy-related cancer networks. Int J Biochem Cell Biol 2011; 43: 1263–6.
Lorin S, Pierron G, Ryan KM, Codogno P, Djavaheri-Mergny M . Evidence for the interplay between JNK and p53-DRAM signalling pathways in the regulation of autophagy. Autophagy 2010; 6: 153–4.
D'Amelio M, Cecconi F . A novel player in the p53-mediated autophagy: Sestrin2. Cell Cycle 2009; 8: 1467.
Yee KS, Wilkinson S, James J, Ryan KM, Vousden KH . PUMA- and Bax-induced autophagy contributes to apoptosis. Cell Death Differ 2009; 16: 1135–45.
Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126: 121–34.
Tasdemir E, M aiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008; 10: 676–87.
Kim KM, Yu TK, Chu HH, Park HS, Jang KY, Moon WS, et al. Expression of ER stress and autophagy-related molecules in human non-small cell lung cancer and premalignant lesions. Int J Cancer 2012; 131: E362–70.
Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell 2007; 25: 193–205.
Hengartner MO . The biochemistry of apoptosis. Nature 2000; 407: 770–6.
Eum KH, Lee M . Crosstalk between autophagy and apoptosis in the regulation of paclitaxel-induced cell death in v-Ha-ras-transformed fibroblasts. Mol Cell Biochem 2011; 348: 61–8.
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G . Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8: 741–52.
Platini F, Perez-Tomas R, Ambrosio S, Tessitore L . Understanding autophagy in cell death control. Curr Pharm Des 2010; 16: 101–13.
Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Souquere S, et al. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem 2006; 281: 30373–82.
Eisenberg-Lerner A, Kimchi A . The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis 2009; 14: 376–91.
White E . Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012; 12: 401–10.
Koneri K, Goi T, Hirono Y, Katayama K, Yamaguchi A . Beclin 1 gene inhibits tumor growth in colon cancer cell lines. Anticancer Res 2007; 27: 1453–7.
Sun Y, Liu JH, Sui YX, Jin L, Yang Y, Lin SM, et al. Beclin1 overexpression inhibitis proliferation, invasion and migration of CaSki cervical cancer cells. Asian Pac J Cancer Prev 2011; 12: 1269–73.
Jiang ZF, Shao LJ, Wang WM, Yan XB, Liu RY . Decreased expression of Beclin-1 and LC3 in human lung cancer. Mol Biol Rep 2012; 39: 259–67.
Roesly HB, Khan MR, Chen HD, Hill KA, Narendran N, Watts GS, et al. The decreased expression of Beclin-1 correlates with progression to esophageal adenocarcinoma: the role of deoxycholic acid. Am J Physiol Gastrointest Liver Physiol 2012; 302: G864–72.
Chen YB, Hou JH, Feng XY, Chen S, Zhou ZW, Zhang XS, et al. Decreased expression of Beclin 1 correlates with a metastatic phenotypic feature and adverse prognosis of gastric carcinomas. J Surg Oncol 2012; 105: 542–7.
Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS, et al. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol 2009; 217: 702–6.
Xie R, Wang F, McKeehan WL, Liu L . Autophagy enhanced by microtubule- and mitochondrion-associated MAP1S suppresses genome instability and hepatocarcinogenesis. Cancer Res 2011; 71: 7537–46.
Narita M, Young AR . Autophagy facilitates oncogene-induced senescence. Autophagy 2009; 5: 1046–7.
Zhuang W, Qin Z, Liang Z . The role of autophagy in sensitizing malignant glioma cells to radiation therapy. Acta Biochim Biophys Sin 2009; 41: 341–51.
Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, Kalkanis SN, et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer 2009; 125: 717–22.
Bareford MD, Park MA, Yacoub A, Hamed HA, Tang Y, Cruickshanks N, et al. Sorafenib enhances pemetrexed cytotoxicity through an autophagy-dependent mechanism in cancer cells. Cancer Res 2011; 71: 4955–67.
Janku F, McConkey DJ, Hong DS, Kurzrock R . Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8: 528–39.
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25: 717–29.
Mathew R, White E . Autophagy, stress, and cancer metabolism: what doesn't kill you makes you stronger. Cold Spring Harb Symp Quant Biol 2011; 76: 389–96.
Eng CH, Abraham RT . The autophagy conundrum in cancer: influence of tumorigenic metabolic reprogramming. Oncogene 2011; 30: 4687–96.
Altman BJ, Jacobs SR, Mason EF, Michalek RD, MacIntyre AN, Coloff JL, et al. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011; 30: 1855–67.
Bhutia SK, Kegelman TP, Das SK, Azab B, Su ZZ, Lee SG, et al. Astrocyte elevated gene-1 induces protective autophagy. Proc Natl Acad Sci U S A 2010; 107: 22243–8.
Shen S, Kepp O, Kroemer G . The end of autophagic cell death? Autophagy 2012; 8: 1–3.
Rosenfeldt MT, Ryan KM . The multiple roles of autophagy in cancer. Carcinogenesis 2011; 32: 955–63.
Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res 2005; 65: 3336–46.
Crazzolara R, Bradstock KF, Bendall LJ . RAD001 (Everolimus) induces autophagy in acute lymphoblastic leukemia. Autophagy 2009; 5: 727–8.
Cao C, Subhawong T, Albert JM, Kim KW, Geng L, Sekhar KR, et al. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res 2006; 66: 10040–7.
Yazbeck VY, Buglio D, Georgakis GV, Li Y, Iwado E, Romaguera JE, et al. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol 2008; 36: 443–50.
Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 2010; 70: 288–98.
Can G, Ekiz HA, Baran Y . Imatinib induces autophagy through BECLIN-1 and ATG5 genes in chronic myeloid leukemia cells. Hematology 2011; 16: 95–9.
Gupta A, Roy S, Lazar AJ, Wang WL, McAuliffe JC, Reynoso D, et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc Natl Acad Sci U S A 2010; 107: 14333–8.
Kamitsuji Y, Kuroda J, Kimura S, Toyokuni S, Watanabe K, Ashihara E, et al. The Bcr-Abl kinase inhibitor INNO-406 induces autophagy and different modes of cell death execution in Bcr-Abl-positive leukemias. Cell Death Differ 2008; 15: 1712–22.
Le XF, Mao W, Lu Z, Carter BZ, Bast RC Jr . Dasatinib induces autophagic cell death in human ovarian cancer. Cancer 2010; 116: 4980–90.
Krause G, Hallek M . On the assessment of dasatinib-induced autophagy in CLL. Leuk Res 2011; 35: 137–8.
Tong Y, Liu YY, You LS, Qian WB . Perifosine induces protective autophagy and upregulation of ATG5 in human chronic myelogenous leukemia cells in vitro. Acta Pharmacol Sin 2012; 33: 542–50.
Evangelisti C, Ricci F, Tazzari P, Chiarini F, Battistelli M, Falcieri E, et al. Preclinical testing of the Akt inhibitor triciribine in T-cell acute lymphoblastic leukemia. J Cell Physiol 2011; 226: 822–31.
Cirstea D, Hideshima T, Rodig S, Santo L, Pozzi S, Vallet S, et al. Dual inhibition of akt/mammalian target of rapamycin pathway by nanoparticle albumin-bound-rapamycin and perifosine induces antitumor activity in multiple myeloma. Mol Cancer Ther 2010; 9: 963–75.
Xiao X, He Q, Lu C, Werle KD, Zhao RX, Chen J, et al. Metformin impairs the growth of liver kinase B1-intact cervical cancer cells. Gynecol Oncol 2012; 127: 249–55.
Tomic T, Botton T, Cerezo M, Robert G, Luciano F, Puissant A, et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis 2011; 2: e199.
Korolchuk VI, Menzies FM, Rubinsztein DC . Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 2010; 584: 1393–8.
Li C, Johnson DE . Bortezomib induces autophagy in head and neck squamous cell carcinoma cells via JNK activation. Cancer Lett 2012; 314: 102–7.
Fels DR, Ye J, Segan AT, Kridel SJ, Spiotto M, Olson M, et al. Preferential cytotoxicity of bortezomib toward hypoxic tumor cells via overactivation of endoplasmic reticulum stress pathways. Cancer Res 2008; 68: 9323–30.
Zhu K . Dunner K Jr, McConkey DJ . Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010; 29: 451–62.
Claerhout S, Lim JY, Choi W, Park YY, Kim K, Kim SB, et al. Gene expression signature analysis identifies vorinostat as a candidate therapy for gastric cancer. PLoS One 2011; 6: e24662.
Yamamoto S, Tanaka K, Sakimura R, Okada T, Nakamura T, Li Y, et al. Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or autophagy-associated cell death in chondrosarcoma cell lines. Anticancer Res 2008; 28: 1585–91.
Hrzenjak A, Kremser ML, Strohmeier B, Moinfar F, Zatloukal K, Denk H . SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J Pathol 2008; 216: 495–504.
Liu YL, Yang PM, Shun CT, Wu MS, Weng JR, Chen CC . Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010; 6: 1057–65.
Park JH, Ahn MY, Kim TH, Yoon S, Kang KW, Lee J, et al. A new synthetic HDAC inhibitor, MHY218, induces apoptosis or autophagy-related cell death in tamoxifen-resistant MCF-7 breast cancer cells. Invest New Drugs 2012; 30: 1887–98.
Lin Z, Bazzaro M, Wang MC, Chan KC, Peng S, Roden RB . Combination of proteasome and HDAC inhibitors for uterine cervical cancer treatment. Clin Cancer Res 2009; 15: 570–7.
Kim J, Guan J, Chang I, Chen X, Han D, Wang CY . PS-341 and histone deacetylase inhibitor synergistically induce apoptosis in head and neck squamous cell carcinoma cells. Mol Cancer Ther 2010; 9: 1977–84.
Zhong JT, Xu Y, Yi HW, Su J, Yu HM, Xiang XY, et al. The BH3 mimetic S1 induces autophagy through ER stress and disruption of Bcl-2/Beclin 1 interaction in human glioma U251 cells. Cancer Lett 2012; 323: 180–7.
Tian S, Lin J, Jun Zhou J, Wang X, Li Y, Ren X, et al. Beclin 1-independent autophagy induced by a Bcl-XL/Bcl-2 targeting compound, Z18. Autophagy 2010; 6: 1032–41.
Lian J, Wu X, He F, Karnak D, Tang W, Meng Y, et al. A natural BH3 mimetic induces autophagy in apoptosis-resistant prostate cancer via modulating Bcl-2-Beclin1 interaction at endoplasmic reticulum. Cell Death Differ 2011; 18: 60–71.
Park KJ, Lee SH, Kim TI, Lee HW, Lee CH, Kim EH, et al. A human scFv antibody against TRAIL receptor 2 induces autophagic cell death in both TRAIL-sensitive and TRAIL-resistant cancer cells. Cancer Res 2007; 67: 7327–34.
Liu N, Tai S, Ding B, Thor RK, Bhuta S, Sun Y, et al. Arsenic trioxide synergizes with everolimus (Rad001) to induce cytotoxicity of ovarian cancer cells through increased autophagy and apoptosis. Endocr Relat Cancer 2012; 19: 711–23.
Kim KW, Moretti L, Mitchell LR, Jung DK, Lu B . Combined Bcl-2/mammalian target of rapamycin inhibition leads to enhanced radiosensitization via induction of apoptosis and autophagy in non-small cell lung tumor xenograft model. Clin Cancer Res 2009; 15: 6096–105.
Shi WY, Xiao D, Wang L, Dong LH, Yan ZX, Shen ZX, et al. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis 2012; 3: e275.
de Medina P, Silvente-Poirot S, Poirot M . Tamoxifen and AEBS ligands induced apoptosis and autophagy in breast cancer cells through the stimulation of sterol accumulation. Autophagy 2009; 5: 1066–7.
Gills JJ, Lopiccolo J, Dennis PA . Nelfinavir, a new anti-cancer drug with pleiotropic effects and many paths to autophagy. Autophagy 2008; 4: 107–9.
Yoo SH, Yoon YG, Lee JS, Song YS, Oh JS, Park BS, et al. Etoposide induces a mixed type of programmed cell death and overcomes the resistance conferred by Bcl-2 in Hep3B hepatoma cells. Int J Oncol 2012; doi: 10.3892/ijo.2012.1585.
Ito S, Koshikawa N, Mochizuki S, Takenaga K . 3-Methyladenine suppresses cell migration and invasion of HT1080 fibrosarcoma cells through inhibiting phosphoinositide 3-kinases independently of autophagy inhibition. Int J Oncol 2007; 31: 261–8.
Li J, Hou N, Faried A, Tsutsumi S, Takeuchi T, Kuwano H . Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Ann Surg Oncol 2009; 16: 761–71.
Liu D, Yang Y, Liu Q, Wang J . Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med Oncol 2011; 28: 105–11.
Tseng HC, Liu WS, Tyan YS, Chiang HC, Kuo WH, Chou FP . Sensitizing effect of 3-methyladenine on radiation-induced cytotoxicity in radio-resistant HepG2 cells in vitro and in tumor xenografts. Chem Biol Interact 2011; 192: 201–8.
Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y . Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 1998; 23: 33–42.
Wu YC, Wu WK, Li Y, Yu L, Li ZJ, Wong CC, et al. Inhibition of macroautophagy by bafilomycin A1 lowers proliferation and induces apoptosis in colon cancer cells. Biochem Biophys Res Commun 2009; 382: 451–6.
Feng R, Wang SY, Shi YH, Fan J, Yin XM . Delphinidin induces necrosis in hepatocellular carcinoma cells in the presence of 3-methyladenine, an autophagy inhibitor. J Agric Food Chem 2010; 58: 3957–64.
Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117: 326–36.
Geng Y, Kohli L, Klocke BJ, Roth KA . Chloroquine-induced autophagic vacuole accumulation and cell death in glioma cells is p53 independent. Neuro Oncol 2010; 12: 473–81.
Cerniglia GJ, Karar J, Tyagi S, Christofidou-Solomidou M, Rengan R, Koumenis C, et al. Inhibition of autophagy as a strategy to augment radiosensitization by the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Mol Pharmacol 2012; 82: 1230–40.
Xu CX, Zhao L, Yue P, Fang G, Tao H, Owonikoko TK, et al. Augmentation of NVP-BEZ235's anticancer activity against human lung cancer cells by blockage of autophagy. Cancer Biol Ther 2011; 12: 549–55.
Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010; 10: 370.
Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A . Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012; 8: 200–12.
Rahim R, Strobl JS . Hydroxychloroquine, chloroquine, and all-trans retinoic acid regulate growth, survival, and histone acetylation in breast cancer cells. Anticancer Drugs 2009; 20: 736–45.
Ng AC . Integrative systems biology and networks in autophagy. Semin Immunopathol 2010; 32: 355–61.
McKnight NC, Jefferies HB, Alemu EA, Saunders RE, Howell M, Johansen T, et al. Genome-wide siRNA screen reveals amino acid starvation-induced autophagy requires SCOC and WAC. EMBO J 2012; 31: 1931–46.
Szyniarowski P, Corcelle-Termeau E, Farkas T, Hoyer-Hansen M, Nylandsted J, Kallunki T, et al. A comprehensive siRNA screen for kinases that suppress macroautophagy in optimal growth conditions. Autophagy 2011; 7: 892–903.
Dengjel J, Hoyer-Hansen M, Nielsen MO, Eisenberg T, Harder LM, Schandorff S, et al. Identification of autophagosome-associated proteins and regulators by quantitative proteomic analysis and genetic screens. Mol Cell Proteomics 2012; 11: M111. 014035.
Dengjel J, Andersen J, Jäättel M, Høyer-Hansen M . Identifying proteins in the autophagosomes by a proteomic approach. Apmis 2008; 116: 424.
Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy 2009; 5: 816–23.
Wu H, Wang F, Hu S, Yin C, Li X, Zhao S, et al. MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts. Cell Signal 2012; 24: 2179–86.
Frankel LB, Wen J, Lees M, Hoyer-Hansen M, Farkas T, Krogh A, et al. microRNA-101 is a potent inhibitor of autophagy. EMBO J 2011; 30: 4628–41.
Behrends C, Sowa ME, Gygi SP, Harper JW . Network organization of the human autophagy system. Nature 2010; 466: 68–76.
Huett A, Xavier R . Building complex biological networks based upon model organisms: mapping the human autophagy interactome via a hybrid yeast-human protein interaction network. Autophagy 2009; 5: 884–6.
Guo H, Ingolia NT, Weissman JS, Bartel DP . Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 2010; 466: 835–40.
Fu LL, Wen X, Bao JK, Liu B . MicroRNA-modulated autophagic signaling networks in cancer. Int J Biochem Cell Biol 2012; 44: 733–6.
Korkmaz G, le Sage C, Tekirdag KA, Agami R, Gozuacik D . miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy 2012; 8: 165–76.
Kovaleva V, Mora R, Park YJ, Plass C, Chiramel AI, Bartenschlager R, et al. miRNA-130a targets ATG2B and DICER1 to inhibit autophagy and trigger killing of chronic lymphocytic leukemia cells. Cancer Res 2012; 72: 1763–72.
Yu Y, Cao L, Yang L, Kang R, Lotze M, Tang D . microRNA 30A promotes autophagy in response to cancer therapy. Autophagy 2012; 8: 853–5.
Shu CW, Liu PF, Huang CM . High throughput screening for drug discovery of autophagy modulators. Comb Chem High Throughput Screen 2012; 15: 721–9.
Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, et al. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One 2009; 4: e7124.
Kimura S, Noda T, Yoshimori T . Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3: 452–60.
Hundeshagen P, Hamacher-Brady A, Eils R, Brady NR . Concurrent detection of autolysosome formation and lysosomal degradation by flow cytometry in a high-content screen for inducers of autophagy. BMC Biol 2011; 9: 38.
Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci U S A 2007; 104: 19023–8.
Farkas T, Hoyer-Hansen M, Jaattela M . Identification of novel autophagy regulators by a luciferase-based assay for the kinetics of autophagic flux. Autophagy 2009; 5: 1018–25.
Shu CW, Madiraju C, Zhai D, Welsh K, Diaz P, Sergienko E, et al. High-throughput fluorescence assay for small-molecule inhibitors of autophagins/Atg4. J Biomol Screen 2011; 16: 174–82.
Acknowledgements
We are grateful to Lei WU, Zi-jie WANG, and Rong SUN for their critical review of this manuscript. This work was supported by the National Natural Science Foundation of China (No 30970643, No 81173093 and No J1103518) and Special Program for Youth Science and Technology Innovative Research Group of Sichuan Province, China (No 2011JTD0026).
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article
Cite this article
Li, X., Xu, Hl., Liu, Yx. et al. Autophagy modulation as a target for anticancer drug discovery. Acta Pharmacol Sin 34, 612–624 (2013). https://doi.org/10.1038/aps.2013.23
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2013.23
Keywords
This article is cited by
-
Selective autophagy in cancer: mechanisms, therapeutic implications, and future perspectives
Molecular Cancer (2024)
-
The Link of Marine Products with Autophagy-Associated Cell Death in Cancer Cell
Current Pharmacology Reports (2019)
-
Autophagy plays a protective role against apoptosis induced by toxicarioside N via the Akt/mTOR pathway in human gastric cancer SGC-7901 cells
Archives of Pharmacal Research (2018)
-
Isoliquiritigenin Induces Cytotoxicity in PC-12 Cells In Vitro
Applied Biochemistry and Biotechnology (2017)
-
Tetrandrine is a potent cell autophagy agonist via activated intracellular reactive oxygen species
Cell & Bioscience (2015)
