
Abstract
We have developed ‘self-oscillating’ gels that undergo spontaneous cyclic swelling–deswelling changes without any on–off switching of external stimuli, as with heart muscle. The self-oscillating gels were designed by utilizing the Belousov–Zhabotinsky (BZ) reaction, an oscillating reaction, as a chemical model of the tricarboxylic acid cycle. We have systematically studied these polymer gels since they were first reported in 1996. Our studies represent innovative research, creating new concepts of functional gels and expanding their potential, and they have attracted attention in many research fields and have inspired related studies. Here, our recent progress on self-oscillating polymer gels is summarized.
Similar content being viewed by others
Introduction
Background
The science and engineering of polymer gels have progressed rapidly since the discovery of volume phase transition phenomena by Tanaka.1 Many types of stimuli-responsive polymer gels that exhibit reversible swelling–deswelling changes in response to environmental changes (for example, solvent composition, temperature, pH change, light, electric field, and so on) have been developed. For example, it is well known that poly(N-isopropylacrylamide) (PNIPAAm) has a low critical solution temperature (LCST) of approximately 32 °C and that these gels undergo an abrupt volume collapse (volume phase transition) when heated to temperatures around the LCST.2 Fundamental studies on these gels have been performed, and their application as functional soft materials, especially as biomaterials or biomimetic materials for actuators (artificial muscle), drug delivery systems (DDS), tissue engineering, purification or separation systems, biosensors, shape memory materials and molecular recognition systems, among others,3, 4, 5, 6, 7, 8, 9, 10 has also been exploited. Furthermore, with the progress of polymer synthesis technology and supramolecular science, gels with unique network structures have been designed to provide rapid response,11, 12 toughness13, 14, 15, 16 and self-healing properties, among other benefits.17, 18
In these ways, many stimuli-responsive polymer and gel systems have been inspired by living systems. However, one of the characteristic and important behaviors in living systems is autonomous oscillation, that is, spontaneous changes with temporal periodicity (called ‘temporal structure’), such as the heartbeat, brain waves, pulsatile secretion of hormones, cell cycle and biorhythms. Nevertheless, there are few studies on polymer and gel systems undergoing self-oscillation under constant conditions without any on–off switching of external stimuli. If such autonomous systems can be realized using completely synthetic polymers, unprecedented biomimetic materials may be created.
Development of ‘self-oscillating’ gel
With this concept in mind, we have developed polymer gels with autonomous function. As one possible method, we designed a coupling system of pH-responsive gels and pH-oscillating reactions. By soaking pH-responsive gels in a solution undergoing a pH-oscillating reaction19 in a continuously stirred tank reactor, autonomous and periodic swelling–deswelling changes of the gel were achieved.20, 21 In this system, however, the oscillating outer solution is created by the continuously stirred tank reactor, and the swelling–deswelling changes of the gel only follow the oscillating outer conditions. Therefore, we further attempted to develop a novel gel that provides mechanical oscillation without external control in a completely closed and non-oscillating outer solution. Eventually, we succeeded in developing such self-oscillating polymer gels by incorporating an oscillating chemical reaction in the polymer network,22 that is, by constructing a built-in circuit of an energy conversion cycle that produced mechanical oscillation within the polymer network itself. In contrast to conventional stimuli-responsive gels, we have developed ‘self-oscillating’ polymer gels that autonomously undergo periodic swelling–deswelling oscillation without on–off switching of external stimuli in a closed solution. In this review, we summarize our recent progress on these self-oscillating polymer gels and their evolution as functional materials.
Design of self-oscillating polymer gels
Oscillating chemical reaction: the Belousov–Zhabotinsky reaction
For the design of the gel, the Belousov–Zhabotinsky (BZ) reaction,23, 24, 25, 26 a well-known oscillating reaction that spontaneously exhibits temporal rhythm and spatial pattern, was exploited. The overall process is the oxidation of an organic substrate (for example, malonic acid (MA) or citric acid) by an oxidizing agent (typically bromate ion) in the presence of a catalyst under acidic conditions. Metal ions or metal complexes with high redox potentials, such as cerium ion, ferroin, or ruthenium tris(2,2′-bipyridine) (Ru(bpy)32+) are widely used as catalysts. While the reaction proceeds, the catalyst undergoes spontaneous redox oscillation, showing periodic changes in the color of the solution under stirring and concentric or spiral wave patterns under stationary conditions. The wave of the oxidized state propagating in the medium is called a ‘chemical wave’. The BZ reaction is often analogized with the TCA cycle, a key metabolic process that takes place in living bodies, and the BZ reaction is recognized as a chemical model for understanding several autonomous phenomena in biological systems.
Mechanism of self-oscillation for gels
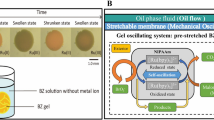
We attempted to convert the chemical oscillation of the BZ reaction into a mechanical change in gels and to generate autonomous swelling–deswelling oscillation under non-oscillatory outer conditions. For this purpose, we prepared a gel composed of PNIPAAm with Ru(bpy)3 covalently bonded to the PNIPAAm network (Figure 1). When the poly(NIPAAm-co-Ru(bpy)3) gel is immersed in catalyst-free BZ solution containing the substrates (MA, NaBrO3 and nitric acid), the reaction occurs in the gel by the catalytic function of the polymerized Ru(bpy)3. The redox changes of the polymerized catalyst moiety (Ru(bpy)32+⇄Ru(bpy)33+) change the volume phase transition temperature of the gel as well as the swelling ratio because the hydrophilicity of the polymer chains increases in the oxidized Ru(III) state and decreases in the reduced Ru(II) state. As a result, the gel exhibits an autonomous swelling–deswelling oscillation with the redox oscillation in the closed solution under constant conditions. We first reported the ‘self-oscillating’ gel in 1996,22 and since then, we have developed many types of biomimetic or smart material systems (Figure 2).27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62

Design concept of self-oscillating gel using the BZ reaction. BZ, Belousov–Zhabotinsky.

Evolution of the self-oscillating polymer gels as functional material systems.
Self-oscillating behavior on several scales
Self-oscillation can be induced on several scales, from the order of polymer chains to bulk gels (Figure 3). In the case of the uncrosslinked linear polymer, the polymer solution exhibits self-oscillations of optical transmittance and viscosity due to spontaneous cyclic soluble–insoluble changes of the polymer. When the gel is much smaller than the chemical wavelength, redox changes occur homogeneously in the gel without pattern formation.28 Then, the swelling–deswelling change of the gel becomes isotropic. However, when the gel size is larger, chemical waves spontaneously evolve and propagate in the gel by coupling with the diffusion of intermediates.29, 30, 31 Then, peristaltic motion of the gel is created. Figure 3c shows the time course of peristaltic motion of cylindrical gel in a solution of BZ substrates.29 The green and orange colors correspond to the oxidized and reduced states of the Ru moiety in the gel, respectively. The chemical waves propagate in the gel at a constant speed in the direction of the gel length. Considering that the orange (Ru(II)) and green (Ru(III)) zones represent simply the shrunken and swollen parts, respectively, the locally swollen and shrunken parts move with the chemical wave, as in the peristaltic motion of living worms or intestines.

Self-oscillation on several scales. (a) Self-oscillating profiles of optical transmittance for poly(NIPAAm-co-Ru(bpy)3) solution. (b) Periodic redox changes of the miniature cubic poly(NIPAAm-co-Ru(bpy)3) gel (lower) and the swelling–deswelling oscillation (upper) at 20 °C. Color changes of the gel accompanied by redox oscillations (orange: reduced state, light green: oxidized state) were converted to 8-bit grayscale (dark: reduced, light: oxidized) by image processing. Transmitted light intensity is expressed as an 8-bit grayscale value. (c) Time course of peristaltic motion of poly(NIPAAm-co-Ru(bpy)3-co-AMPS) gel in a solution of BZ substrates at 18 °C. The green and orange colors correspond to the oxidized and reduced states of the Ru moiety in the gel, respectively. AMPS, 2-acrylamido-2′-methylpropanesulfonic acid; BZ, Belousov–Zhabotinsky.
Control of self-oscillating chemomechanical behaviors
Concentration- and temperature-dependence of oscillation
The oscillating behavior of the BZ reaction changes depending on concentration and temperature as characteristics of the chemical reaction. Typically, the oscillation period increases with a decrease in the initial concentration of substrates. Furthermore, in general, the oscillation frequency (the reciprocal of the period) of the BZ reaction tends to increase as the temperature increases, in accordance with the Arrhenius equation. The swelling–deswelling amplitude of the gel increases with an increase in the oscillation period and amplitude of the redox potential changes.28 Therefore, the swelling–deswelling amplitude of the gel is controllable by changing the initial concentration of the substrates as well as the temperature.
Control of self-oscillating behaviors by designing the chemical structure of the gel
The self-oscillating behaviors can also be controlled by designing the chemical and physical structures of the gels. There are several variations of the gels with different chemical components. For example, it was found to be effective to copolymerize AMPS (2-acrylamido-2′-methylpropanesulfonic acid) with a poly(NIPAAm-co-Ru(bpy)3) gel network to generate a large amplitude of volume change of the gel because the gel had a microphase-separated structure due to poor solvation during the polymerization process (Figure 4a).32

(a) Chemical structure of poly(NIPAAm-co-Ru(bpy)3-co-AMPS) gel and an SEM micrograph of the gel. (b, c) Chemical structures of poly(NIPAAm-co-Ru(bpy)3-co-AMPS-co-MAPTAB) gel and poly(EMAAm-co-Ru(bpy)3) gel. (d) Chemical structure of poly(NIPAAm-co-Ru(bpy)3-co-APMA) microgel and the swelling–deswelling oscillating behavior of the macrogel prepared by assembling and crosslinking the microgels. AMPS, 2-acrylamido-2′-methylpropanesulfonic acid; GA, glutaric dialdehyde; SEM, scanning electron microscope.
The operating conditions for the self-oscillation are limited to conditions under which the BZ reaction occurs. For potential application as functional bio- or biomimetic materials, it is necessary to design a self-oscillating polymer that acts under biological conditions. To induce self-oscillation of polymer systems under physiological conditions, BZ substrates other than organic ones (MA, citric acid, and so on) must be built into the polymer system itself. For this purpose, we have synthesized a quaternary copolymer that includes both pH-control and oxidant-supplying sites in the poly(NIPAAm-co-Ru(bpy)3) chain at the same time33 (Figure 4b). Using this polymer, self-oscillation was actually observed with the addition of only the organic acid (that is, MA).
To induce self-oscillation while maintaining a larger amplitude at higher temperatures and at physiological body temperatures for potential use as biomaterials, we prepared a self-oscillating gel composed of a thermosensitive EMAAm (N,N′-ethylmethylacrylamide) polymer exhibiting a higher LCST than that of PNIPAAm34 (Figure 4c). The self-oscillating behavior of the poly(EMAAm-co-Ru(bpy)3) gel was investigated by comparing it with gels composed of a thermosensitive NIPAAm polymer with a lower LCST and with non-thermosensitive DMAAm (N,N′-dimethylacrylamide) polymer. The design concept of self-oscillation at higher temperatures without a decrease in swelling–deswelling amplitude was demonstrated by utilizing a thermosensitive polymer exhibiting a higher LCST.
Remarkable swelling–deswelling changes by assembled self-oscillating microgels
Several strategies have been used to improve the response of gels, such as introducing porous microstructures and decreasing the size of the gels. In addition, in living muscle, a hierarchical structure exists to amplify the microscopic movements of actin–myosin to macroscopic displacements. Such a hierarchical structure was introduced into the self-oscillating gel to obtain a large amplitude of swelling–deswelling oscillation. Soft actuators assembled from self-oscillating submicron-sized particles (microgels) were fabricated35 (Figure 4d). First, microgels composed of NIPAAm, Ru(bpy)3 and APMA (N-(3-aminopropyl) methacrylamide hydrochloride) were prepared by aqueous free-radical precipitation polymerization. Then, the microgels were assembled and crosslinked using glutaric dialdehyde to induce a chemical reaction between the amino groups located on the microgels’ exterior. The resulting macrogels exhibited large displacements due to the cooperative dispersing/flocculating motion of the constituent microelements.
Comb-type self-oscillating gel
Another effective method of improving the response of the gels is to introduce grafted side chains with freely mobile ends on the backbone networks of the gels. For example, comb-type grafted PNIPAAm gels were shown to exhibit a fast deswelling response to temperature change by the guiding effect of hydrophobic interaction between side chains that have freely mobile ends.11, 12 These network-arranged designs are expected to be applicable for improving the swelling–deswelling kinetics of the redox change in the self-oscillating gel.
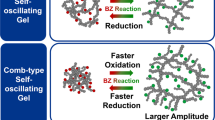
We designed a novel comb-type self-oscillating gel (MSG) that has Ru(bpy)3 on both the main and side chains (Figure 5).36 Unlike the conventional type of self-oscillating gels (MG) in which Ru(bpy)3 is immobilized only on the main chains, faster swelling–deswelling changes are expected because the grafted side chains with freely mobile ends can respond to the redox change of Ru(bpy)3 more quickly than the main chains. As a result, self-oscillation with a larger swelling–deswelling amplitude can be achieved.

Chemical structure of comb-type poly(NIPAAm-co-Ru(bpy)3)-grafted poly(NIPAAm-co-Ru(bpy)3) gel and self-oscillating behaviors for conventional poly(NIPAAm-co-Ru(bpy)3) gel and the comb-type gel at 20 °C in the BZ substrate solution containing NaBrO3, MA, and HNO3. BZ, Belousov–Zhabotinsky; MA, malonic acid.
Figure 5 shows the self-oscillating behaviors of MG and MSG in the catalyst-free BZ solution. Although the period was almost the same (∼550 s), the MSG exhibited oscillation with faster and larger size changes than the MG. The volume change ratio was 31% for MSG, which is much larger than the 7.1% for MG. This increased swelling–deswelling amplitude is due to faster kinetics in the MSG. During oscillation, the deswelling rate of MSG was 5.4 times faster than that of MG, and the swelling rate of MSG was 7.8 times faster than that of MG. Fast responses to the Ru(bpy)3 redox oscillation caused the large amplitude of mechanical oscillation. The smaller amplitude for MG may be because the oxidation or reduction process in the next cycle starts before the gel sufficiently deswells or swells. Thus, by introducing the self-oscillating grafted chains, mechanical oscillation with a larger amplitude was achieved.
On–off regulation of self-oscillation by external stimuli
On–off regulation of self-oscillation by external stimuli such as temperature change,37 addition and removal of organic acid,38 and photo-irradiation is also possible.39, 40, 41 In particular, Ru(bpy)3 is photosensitive in the reduced state. Photo-irradiation either causes the other reaction pathway to stop or causes oscillation by producing an intermediate to act as an inhibitor (Br−) or an activator (HBrO2), depending on the composition of the BZ solution.26 By utilizing such photochemical characteristics of the BZ reaction, photo-regulation of the self-oscillating motion of the gel was experimentally demonstrated.39, 40 Balazs et al.63, 64 theoretically demonstrated light-guided motility of the gel by model simulation.
In this photo-regulation system utilizing the photosensitivity of Ru(bpy)3, there is no difference between the photo-irradiated and non-irradiated conditions in the hydrated state of the polymer chains (and consequently, in the swelling state of the gel). However, we have designed photo-regulated self-oscillating systems based on hydration and dehydration changes of the polymer by photo-irradiation. For this purpose, spirobenzopyran was introduced into the poly(NIPAAm-co-Ru(bpy)3) as a photochromic site41 (Figure 6). Even under the acidic conditions necessary for the BZ reaction, photochromism of the spirobenzopyran occurs, and the LCST of the polymer solution shifts to a lower temperature with isomerization from McH to spirobenzopyran by photo-irradiation.

(Upper) Chemical structure of poly(NIPAAm-co-Ru(bpy)3-co-Sp) (PNRS). (Lower) Oscillating profile of the PNRS aqueous solution containing BZ substrates (MA, NaBrO3 and HNO3) at 21.5°C. (a) In the dark; (b) in response to on–off switching of photo-irradiation. BZ, Belousov–Zhabotinsky; MA, malonic acid.
When the polymer is dehydrated by photo-irradiation, the BZ reaction stops because the dehydrated polymer enters the globule state and inhibits the contact between the ruthenium catalyst and the BZ substrates. Figure 6 shows the oscillating profile of optical transmittance for the PNRS aqueous solution containing the BZ substrates in response to on–off switching by photo-irradiation (irradiation period: from 370 to 670 s). A wavelength of 570 nm, at which there is an isosbestic point for the redox states, was selected to detect the signal derived from the coil–globule oscillation of the polymer. The oscillation stopped after photo-irradiation. The reaction stops because the contact between the polymerized catalyst and the reactants dissolved in the solution is suppressed when the polymer becomes hydrophobic and insoluble. The observed decrease in optical transmittance after photo-irradiation suggests that the oscillation stops due to the aggregation of hydrophobic polymer chains. After turning the light off, the transmittance gradually increased, showing smaller amplitude compared with that before photo-irradiation. Once the polymer aggregates, it might be difficult to rehydrate it back to the initial soluble state. In fact, aggregated polymers were observed in the experiment. The small amplitude is attributed to the slow hydration of the polymer chains. This result suggests that the on–off switching of self-oscillation was achieved predominantly by the change in polymer solubility due to photochromic isomerization from McH to spirobenzopyran, not because of the photosensitivity of Ru(bpy)3. Such a system enables different temporal and spatial photo-regulation of self-oscillation from the previous photo-regulation systems. Theoretical simulation of chemomechanical behaviors corresponding to this photo-regulation system was also demonstrated by Balazs, et al.65
Design of biomimetic soft actuators
Ciliary motion actuator (artificial cilia), self-walking gel, self-propelled motion, composite gel
By exploiting such macroscopic swelling–deswelling oscillation of the gel, novel biomimetic actuators can be created. As applications to autonomous biomimetic actuators, ciliary motion actuators (artificial cilia)42, 43 and self-walking gels,44 which move forward by repeated autonomous bending and stretching like a living looper, were realized (see Figure 2). Furthermore, the self-propelled motion of these gels was theoretically and experimentally demonstrated.45 A cylindrical self-oscillating gel with a gradient in crosslinking density was prepared by photopolymerization. To introduce the gradient structure, during the gelation, one side of the pre-gel solution in a glass capillary was irradiated with light. The resulting gel has denser crosslinking at the photo-irradiated side and bends toward that direction in the reduced state. In the catalyst-free BZ solution, the gel repeated the bending and stretching motion, and the position of the center of the gel appeared to shift slowly upward (toward the region with lower crosslinking density).
Control of the motion by preparing a composite gel made of self-oscillating and nonactive gels was also attempted.46 The disk-shaped self-oscillating gels were arranged around the corners of a polygonal sheet of poly(acrylamide) (PAAm) gel. Wave propagation was controlled by changing the patch size, the catalyst content of the BZ gel, and the spacing between the patches.
Theoretical simulation of the self-oscillating gel
These self-oscillating gels are unique material systems containing reaction-diffusion and chemomechanical motion. Because the chemical reaction and the mechanical motion affect each other with feedback control, the oscillating behavior of the gels becomes more complicated compared with the swelling–deswelling behavior of conventional stimuli-responsive gels. Thus, theoretical simulation is an effective tool for understanding these chemomechanical behaviors. Balazs et al.63, 64, 65, 66, 67, 68, 69 developed a mathematical model for simulating the chemomechanical behaviors of the self-oscillating gels. Since their first report in 2006,66 Balazs et al. have demonstrated several aspects of the self-oscillating behavior of the gels by theoretical simulation. Many interesting phenomena and possible actuating behaviors, some of which have been experimentally realized, have been demonstrated theoretically. The investigations from further theoretical studies will provide helpful guidelines for robotic applications, understanding living phenomena, and so on.
Design of autonomous mass transport systems
Self-driven gel conveyer: autonomous transportation on the self-oscillating gel surface by peristaltic motion
To realize a self-driven gel conveyer as a novel autonomous mass transport system, we attempted to transport an object by utilizing the peristaltic motion of the self-oscillating gel.32, 47, 48, 49 A model object, such as a cylindrical or spherical PAAm gel, was put on the surface of a self-oscillating gel sheet. It was observed that the object was transported on the gel surface with the propagation of the chemical wave as it rolled (see the photograph in Figure 2). The velocity and the inclination angle of the chemical wave were changed by changing the concentration of the outer solution. The ability of the cylindrical gel to be transported was estimated for controlled chemical waves with several inclination angles and velocities. The cylindrical PAAm gel was not transported when the inclination angle was less than approximately 3°. The mass transportability did not depend on the velocity of the chemical wave, but on the diameter of the cylindrical PAAm gel and the inclination angle of the wave front. To analyze this result, we proposed a model to describe the mass transport phenomena based on the Hertz contact theory, and the relationship between the transportability and the peristaltic motion was investigated. Calculations using the theoretical equation indicated that the minimum inclination angle was 3°, the same as the angle determined from the experiment. The model supported the idea that the sheer wave front of the peristaltic motion was necessary to transport the cylindrical gels.
Then, a surface figure capable of transporting microparticles in one direction was designed to fabricate a more versatile self-driven gel conveyer. A self-oscillating gel with a grooved surface was fabricated using a PDMS template, and the effectiveness of the surface design was investigated. Poly(AAm-co-AMPS) gel beads with diameters of several hundred microns to several millimeters were transported on the grooved surface of the self-oscillating gel by its autonomous peristaltic motions.47 The traveling direction of the peristaltic motion could be confined to the direction along the grooves by designing the groove distance to be shorter than the wavelength of the chemical wave. Consequently, several gel beads were transported in parallel.
The influence of these surface properties on the transport behaviors has also been investigated.48 Instead of PAAm gel, several types of copolymer gels consisting of AAm with AMPS, N-(3-aminopropyl)methacrylamide hydrochloride (APMA), N-(hydroxymethyl)acrylamide, and MMA (methyl methacrylate) were prepared as model cargos with different surface properties: positive or negative charge and hydrophilicity or hydrophobicity, respectively. The adhesion force (because of their hydrophobic interaction) between the cylindrical poly(AAm-co-MMA) gel and the self-oscillating gel sheet was too strong to apply the transport model. For the other gels, the adhesive force is not sufficient to prevent transportation, which agrees with predictions from swelling, zeta potential and contact angle measurements. Next, the effect of surface roughness on the transportation was investigated. Higher surface roughness was shown to be more effective for transporting the loaded gel because the frictional force increases and the moment of force of the rotational motion increases.
Autonomous intestine-like motion of a tubular self-oscillating gel
Furthermore, to construct autonomous mechanical pumping systems such as intestines, we fabricated a self-oscillating gel in a tubular shape50 (Figure 7). The tubular self-oscillating gels were fabricated by photopolymerization. Several types of tubular self-oscillating gels that exhibit autonomous peristaltic motion were prepared. First, a tubular poly(NIPAAm-co-Ru(bpy)3) gel adhered to the inner wall of a glass capillary was prepared, and the periodic inner diameter changes during the BZ reaction were analyzed. Second, by removing the gel from the glass capillary, a tubular gel that could swell and deswell freely without a mechanical restraint was prepared (Figure 7b). Then, a tubular gel with an interpenetrating network structure composed of self-oscillating and non-oscillating polymers was prepared. These tubular self-oscillating gels exhibited various types of peristaltic motion.

(a) Schematic illustration of autonomous mass transport by peristaltic pumping of a tubular self-oscillating gel. (b) Time course images of the peristaltic motion of the tubular poly(NIPAAm-co-Ru(bpy)3-co-AMPS) gel. (c) Behavior of the autonomous transport of a CO2 bubble in the gel tube by peristaltic pumping. (d) Change in the position of the bubble. (e) The velocity of the bubble. AMPS, 2-acrylamido-2′-methylpropanesulfonic acid.
In addition, it was demonstrated that an object could be autonomously transported in the gel tube by the peristaltic pumping motion, which was similar to that of an intestine. Figure 7c shows the behavior of a CO2 bubble in the tubular poly(NIPAAm-co-Ru(bpy)3) gel. When the bubble became large enough to contact the inner surface of the gel layer, it started to move intermittently by repeated deformation and restoration in the direction of the chemical wave propagation. When the chemical wave reached the contact point, the bubble was squashed and deformed by swelling of the gel layer at that point. Then, the bubble was mechanically pushed forward by the peristaltic pumping mechanism. After the wave passed through, the gel layer deswelled, and the squashed bubble returned back to its initial round shape. Due to the decrease in pushing force and negative pressure, the bubble moved backward slightly. After that, the movement of the bubble stopped for a while. Thus, the movement was intermittent. By repeating this process, the bubble was transported in the gel tube.
Figure 7d and e shows the changes in the position and velocity of the bubble, respectively. The velocity was calculated by differentiating the position with respect to time. It is obvious that a net movement of the bubble occurs by repeating backward and forward movements. Potential applications in artificial intestines, artificial digestive tracts, and so on can be envisioned. Furthermore, there is the possibility of autonomous flow of an inner fluid. We are investigating an application for a novel micropump for microfluidic systems.
Self-oscillating polymer brushes
Recently, surface modification techniques for polymer chains have progressed a great deal with the development of new polymer synthesis methods. In particular, SI-ATRP (surface-initiated atom transfer radical polymerization) is one of the most effective modification methods for preparing a well-defined dense polymer brush structure, or polymer brush, on substrates. Thus, a self-oscillating polymer brush prepared by SI-ATRP can be expected to create a new self-oscillating surface with autonomic function, such as cilia, which will lead to potential applications in transporting systems for nanomaterials or flow control in microfluidics.
We prepared self-oscillating polymer brushes on glass substrates through SI-ATRP51 (Figure 8). The self-oscillating polymer was grafted onto the inner surface of a glass capillary. Figure 8b shows an image observed with fluorescence microscopy, which shows that the self-oscillating polymer brush was successfully grafted onto the inner surface of the capillary. Then, the catalyst-free BZ solution was fed into the capillary, and the BZ reaction on the inner surface was observed by fluorescence microscopy. Spatiotemporal image analyses were performed in different locations (1–4) of the glass capillary, and the oscillating profiles of the fluorescence intensity were compared (Figure 8c). Oscillations occurred at each position with a phase difference. This finding suggests that the chemical wave propagated in the self-oscillating polymer brush layer on the inner surface of the glass capillary. A self-oscillating surface to generate spontaneous periodic changes was demonstrated using synthetic polymers as a novel autonomous functional surface, which has potential applications in systems such as nano-transport systems.

Preparation of self-oscillating polymer brush by surface-initiated atom transfer radical polymerization (SI-ATRP). (a) Chemical structure of the self-oscillating polymer brush. (b) Images of glass capillary modified with the self-oscillating polymer observed by fluorescence microscopy. (c) Oscillating profile of fluorescence intensity at each position for the self-oscillating polymer brush on the inner surface of the glass capillary. The solution containing MA, NaBrO3 and HNO3 is enclosed in the capillary at 25 °C. MA, malonic acid.
Self-oscillating fluids
Transmittance and viscosity oscillation of a polymer solution and microgel dispersion
In the case of the uncrosslinked linear polymer, as shown in Figure 2, the polymer (poly(NIPAAm-co-Ru(bpy)3) undergoes spontaneous cyclic soluble–insoluble changes, and the transmittance of the polymer solution oscillates autonomously with redox changes of the copolymerized Ru(bpy)3.52 Furthermore, we prepared submicron-sized poly(NIPAAm-co-Ru(bpy)3) gel beads by surfactant-free aqueous precipitation polymerization and analyzed the oscillating behavior of the microgel dispersions.53, 54, 55, 56 The microgel dispersion also exhibited transmittance oscillation due to swelling–deswelling changes of the microgels. With increasing temperature, the oscillation period decreased and the amplitude increased slightly. When the temperature increased to near the volume phase transition temperature of the reduced microgel, the microgels showed dispersing/flocculating oscillation as well as swelling–deswelling oscillation (see Figure 2) because the reduced and shrunken microgels lost colloidal stability, which resulted in a remarkable increase of amplitude in the optical transmittance oscillation. Due to such behaviors, as mentioned before, the macroscopic self-oscillating gel prepared by crosslinking the assembled microgels exhibits a faster response and leads to a larger amplitude of swelling–deswelling oscillation.35
In the cases of both the polymer solution and the microgel dispersion, viscosity oscillation was observed with optical transmittance oscillation.57, 58, 59 In the microgel dispersion, it was found that the viscosity oscillation exhibits two different waveforms; a simple pulsatile waveform and a complex waveform with two peaks per period. The difference in waveform is due to the difference in the oscillating behavior of the microgels: swelling/deswelling or dispersing/flocculating oscillation. The oscillating behavior can be controlled by changing the concentration of microgels, Ru(bpy)3 contents, crosslinking density, and so on. It is expected that these polymer solutions and microgel dispersions can be applied as novel functional fluids.
Autonomous viscosity oscillation by reversible complex formation of terpyridine-terminated branched-PEG in the BZ reaction
Recently, we realized autonomous viscosity oscillations of a polymer solution based on different mechanisms;60, 61 that is, autonomous viscosity oscillations of polymer solutions coupled with metal–ligand association/dissociation between Ru and terpyridine (tpy), driven by the BZ reaction. The tpy ligand for the Ru catalyst was attached to the terminals of poly(ethylene glycol) (PEG) with different numbers of branches (linear-, tetra- and octa-PEG) (Figure 9). It is well known that mono-tpy coordination is stable when Ru is oxidized (Ru(tpy)3+), whereas bis-tpy coordination is stable when the Ru center is reduced (Ru(tpy)22+).70 In the oxidized state, these polymers existed as solutions. In contrast, when the Ru center was reduced, gels were obtained for the tetra- and octa-PEG owing to the formation of a three-dimensional polymer network through Ru–tpy coordination (Figure 9). By increasing the number of PEG branches, we succeeded in decreasing the number of crosslinking points necessary for gelation. This was qualitatively explained by the tree-like structure theory stating a rate of reaction of crosslinking points for a branched polymer at the gelation point. Furthermore, the gelation kinetics of the octa-PEG system were approximately four times faster than those of the tetra-PEG system.

Difference in the appearance of 2-, 4- and 8-branched PEGs at equilibrium depending on the redox state of the modified Ru metal ligand at the terminal. PEG, poly(ethylene glycol).
The polymer solutions exhibited self-oscillation of absorbance and viscosity when BZ substrates were added to the solutions of Ru2+-tpy-modified tetra-/octa-PEG. This indicated that the Ru(tpy)22+ attached to the polymer ends could serve as a metal catalyst for the BZ reaction. The viscosity oscillation profiles of the tetra- and octa-PEG were compared under the optimized conditions for each system (Figure 10). As expected, the octa-PEG was much more effective than the tetra-PEG system in providing a large oscillation amplitude as well as a higher baseline viscosity. For the octa-PEG, the baseline viscosity and the amplitude were approximately twice as high and 10 times larger, respectively, than for the tetra-PEG system. This is likely because the number of crosslinking points necessary for gelation was decreased by increasing the branch number of PEG, as predicted from the tree-like structure theory. As a result, the maximum value of crosslinking points in oscillation is closer to that necessary for gelation. Thus, viscosity oscillation occurred in the region of higher viscosity with a larger amplitude.

Oscillating profiles of viscosity of the aqueous solution containing Ru(tpy)2-tetra or Ru(terpy)2-octa-PEG, HNO3, NaBrO3 and MA at 25 °C. MA, malonic acid; PEG, poly(ethylene glycol).
Self-oscillating micelles
Furthermore, we prepared a novel block copolymer that could undergo spontaneous unimer–micelle oscillation under constant conditions.62 Block copolymers generally create static stable structures, such as micelles, by self-assembly. Thus far, most studies involving block copolymers have focused on obtaining the required static structure by controlling the self-assembly. The introduction of dissipative structures into block copolymer systems would provide the novel concept of polymer self-organization with the ability to undergo rhythmic structural change. This type of system can be regarded as self-organization in which self-assembly and dissipative structure assist each other. We demonstrated dynamic self-assembly of a block copolymer driven by the dissipative structure of the BZ reaction. The target diblock copolymer (PEO-b-P(NIPAAm-r-Ru(bpy)3) was successfully prepared by RAFT random copolymerization of NIPAAm and a vinyl monomer with a Ru(bpy)3 side chain from poly(ethylene oxide) (PEO)-based macro-CTA (Figure 11).

(Upper) Chemical structure of PEO-b-P(NIPAAm-r-Ru(bpy)3). (Lower) Left: the concept of the self-oscillating micelles, right: hydrodynamic radius (Rh) of the diblock copolymer in the reduced and oxidized states of the Ru(bpy)3 side chain as a function of temperature. CMT, critical micellization temperature; PEO, poly(ethylene oxide).
The aggregation behavior of the PEO-b-P(NIPAAm-r-Ru(bpy)3) diblock copolymer was first investigated in both reduced (PEO-b-P(NIPAAm-r-Ru(bpy)32+)) and oxidized (PEO-b-P(NIPAAm-r-Ru(bpy)33+)) states at the same ionic strength. The average hydrodynamic radius (Rh) increased sharply above a certain temperature, indicating that self-assembly from unimer to micelle occurs at a specific temperature irrespective of the redox state of Ru(bpy)3. The Rh values at lower temperatures were approximately 7–10 nm, suggesting single polymer chain dissolution, whereas those at higher temperatures ranged between several 10s of nm to more than 100 nm. The reversibility of the unimer–micelle transition with respect to the temperature was also confirmed. More importantly, an approximately 2.5 °C difference in aggregation temperature was observed between the oxidized and reduced states. In the reduced state, the diblock copolymer started to aggregate above 25.6 °C, whereas the oxidized polymer aggregated above 28.1 °C. This is because the hydrophilicity of the second block increased due to the increase in charge. Using this difference in the transition temperatures, a dynamic unimer–micelle transition under the conditions of the BZ reaction was demonstrated.
By adding the BZ substrates (NaBrO3, HNO3 and MA) into the block copolymer solution, the oscillating behavior of the diblock copolymer under constant conditions was analyzed. The scattering intensity and Rh were measured as a function of time with the aid of time-resolved dynamic light scattering (Figure 12). Rhythmic oscillation of the Tyndall effect was observed accompanying the color changes between orange and green based on the characteristic BZ redox oscillation of Ru(bpy)3. When the color of the solution was orange, intense scattering of incident laser light was observed. In contrast, the Tyndall effect weakened in the oxidized state because the aggregation temperature of the oxidized polymer was higher than the experimental temperature. This qualitative observation strongly implied that the diblock copolymer solution passed between two different stages of unimer (oxidized form) and micelle (reduced form) that were sufficiently large to scatter light.

(Upper) Photographs of the block copolymer solution showing periodic changes in the Tyndall effect derived from the self-oscillating micelle synchronized with the BZ reaction. The sample was irradiated with a red laser from the left-hand side. (Lower) (a) Normalized scattering intensity of the block copolymer solution as a function of time. (b) Schematic illustration of unimer–micelle oscillation and the oscillating profiles of the Rh of the diblock copolymer determined by time-resolved DLS measurements. BZ, Belousov–Zhabotinsky; DLS, dynamic light scattering.
Figure 12a shows the oscillating profiles of the scattering intensity for the 0.5 wt% block copolymer solution at a temperature (26.0 °C) between the aggregation temperatures of the reduced and oxidized states. The baseline of the scattering intensity increased with time, indicating that the size of the micelles essentially increased during the oscillation process. Figure 12b shows the oscillating profiles of the average Rh calculated from the dynamic light scattering measurements. The Rh value can be calculated from the fitting of the autocorrelation function, which was collected every 2 s. Figure 12 clearly shows that the size of the scattering particles rhythmically oscillates in synchrony with the BZ reaction. The Rh baseline increased with time, suggesting continuous micellar growth during oscillation, as estimated from the scattering oscillation profiles. From these results, it was suggested that the reduced block copolymer remained even when the solution was in the oxidized state. In the oxidized state of the diblock copolymer, even the smallest average Rh value of a scatterer was estimated to be 17 nm. This value seemed too large to indicate that the micelles were completely broken down into individual polymer chains. Scattering intensity is theoretically proportional to the sixth power of particle size and is proportional to particle number. In the presence of the micelles, the dynamic light scattering signal is heavily weighted by the larger component even if the amount of that component is small.
Thus, we have reported a novel block copolymer that could undergo spontaneous unimer–micelle oscillation under constant conditions without the application of any on–off switching of external stimuli. There have been numerous studies on block copolymers in the area of controlled self-assembly, focused mainly on the formation of static structures under equilibrium conditions. However, this is the first report of a synthetic block copolymer that exhibits ‘temporal structure,’ undergoing self-oscillation between unimer and micelle, as an example of a dissipative structure. This result provides a novel concept for self-organization of block copolymers by combining self-assembly with dissipative structures.
The BZ reaction in protic ionic liquids
Furthermore, we realized the BZ reaction using hydrated protic ionic liquid (PIL) as a solvent for the first time (Figure 13).71 Certain types of PILs containing hydrogen sulfate or methane sulfonate anion have been found to cause self-oscillation without adding any strong acid, such as nitric or sulfuric acid, which commonly acts as a proton source for the BZ reaction when Ru(bpy)3, NaBrO3 and MA are mixed in water. The requirements of PILs for the BZ reaction are as follows: (a) it must be water miscible, (b) anion is not the conjugate base of a super-strong acid and (c) the cation structure does not possess an allyl group. In the case of diethylmethylammonium hydrogen sulfate [dema-H+][HSO4−] aqueous solution, stable self-oscillation continued for as long as 7 h while maintaining a large amplitude under optimized conditions. The pKa of [dema-H+][HSO4−] was estimated to be 0.31, which suggests that the [dema-H+][HSO4−] is a weaker acid than nitric acid (pKa=−1.8) or sulfuric acid (pKa=−3). Through the oscillatory [dema-H+][HSO4−] concentration region (0.3–3.0 M), the pH of [dema-H+][HSO4−] aqueous solution remained between 1.82 and 2.41. These pH values are much higher than the pH value of 0.3 M HNO3 (pH=0.52), which means that the BZ reaction is possible at a higher pH with PILs. The observed redox oscillation in the PIL solutions exhibited a shorter period than that in the conventional HNO3 solution. From the analysis of the observed waveforms based on the chemical models for the BZ reaction (FKN mechanism), it is suggested that neutral [dema] derived from [dema-H+] cation behaves as a proton acceptor to provide rapid proton production accompanying the reduction reaction from Ru(bpy)33+ to Ru(bpy)32+, which results in a short oscillation period, as well as stable and long-lasting oscillation. Investigation of more optimized ion structures for a stable and long-lasting BZ reaction under mild conditions is now in progress for evolution of the self-oscillating gels.

Self-oscillation profile of the BZ reaction in (a) [dema-H+][HSO4−] (1.3 M) and (b) HNO3 (0.3 M) in aqueous solution. BZ, Belousov–Zhabotinsky.
Future prospects
As mentioned above, we developed novel ‘self-oscillating’ polymer gels that exhibit autonomous swelling–deswelling oscillation similar to those of heart muscle under constant external conditions. Since the first report in 1996,22 we have systematically studied these polymer gels. In this review, our recent progress on the self-oscillating polymer gels as functional materials has been summarized. As an innovative study to propose a new concept of functional gels and to expand the potential of these gels, our studies have attracted much attention in many research fields of polymer science, materials science, physical chemistry, robotics, theoretical simulation, biophysics and others. Many similar and applied studies that follow the same mechanism will be possible. In fact, it now seems that our studies have inspired various similar works.72 We will continue to advance our research toward more innovations.
References
Tanaka, T. Gels. Sci. Am. 244, 124–136 (1981).
Schild, H. G. Poly(N-isopropylacrylamide): experiment, theory and application. Prog. Polym. Sci. 17, 163–249 (1992).
Yoshida, R. Design of functional polymer gels and their application to biomimetic materials. Curr. Org. Chem. 9, 1617–1641 (2005).
Lendlein, A. & Shastri, V. P. Stimuli-sensitive polymers. Adv. Mater. 22, 3344–3347 (2010).
Urban, M. W. (ed.) Handbook of stimuli-responsive materials (Wiley-VCH, Weinheim, 2011).
Bhattacharyya, D. & Schafer, T. (eds) Responsive membranes and materials (John Wiley & Sons, Ltd., West Sussex, UK, 2013).
Ottenbrite, R. M., Park, K., Okano, T. & Peppas, N. A. (eds) Biomedical Applications of Hydrogels Handbook (Springer, New York, 2010).
Yang, J., Yamato, M., Sekine, H., Sekiya, S., Tsuda, Y., Ohashi, K., Shimizu, T. & Okano, T. Tissue engineering using laminar cellular assemblies. Adv. Mater. 21, 3404–3409 (2009).
Miyata, T. In Supramolecular Design for Biological Applications (ed. Yui, N.) pp 191–225 (CRC Press, Boca Raton, 2002).
Hoffman, A. S. Stumuli-responsive polymers: biomedical applications and challenges for clinical translation. Adv. Drug. Deliv. Rev. 65, 10–16 (2013).
Yoshida, R., Uchida, K., Kaneko, Y., Sakai, K., Kikuchi, A., Sakurai, Y. & Okano, T. Comb-type grafted hydrogels with rapid de-swelling response to temperature changes. Nature 374, 240–242 (1995).
Kaneko, Y., Sakai, K., Kikuchi, A., Yoshida, R., Sakurai, Y. & Okano, T. Influence of freely mobile grafted chain length on dynamic properties of comb-type grafted poly(N-isopropylacrylamide) hydrogels. Macromolecules 28, 7717–7723 (1995).
Gong, J. P., Katsuyama, Y., Kurosawa, T. & Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 15, 1155–1158 (2003).
Okumura, Y. & Ito, K. The polyrotaxane gel: a topological gel by figure-of–eight cross-links. Adv. Mater. 13, 485–487 (2001).
Haraguchi, K. & Takehisa, T. Nanocomposite hydrogels: a unique organic-inorganic network structure with extraordinary mechanical, optical, and swelling/de-swelling properties. Adv. Mater. 14, 1120–1124 (2002).
Sakai, T., Akagi, Y., Matsunaga, T., Kurakazu, M., Chung, U. & Shibayama, M. Highly elastic and deformable hydrogel formed from tetra-arm polymers. Macromol. Rapid Commun. 31, 1954–1959 (2010).
Wang, Q., Mynar, J. L., Yoshida, M., Lee, E., Lee, M., Okuro, K., Kinbara, K. & Aida, T. High-water-content mouldable hydrogels by mixing clay and a dendritic molecular binder. Nature 463, 339–343 (2010).
Harada, A. & Takashima, Y. Macromolecular recognition and macroscopic interactions by cyclodextrins. Chem. Rec. 13, 420–431 (2013).
Rabai, G., Kustin, K. & Epstein, I. R. A systematically designed pH oscillatior: the hydrogen peroxide-sulfite-ferrocyanide reaction in a continuous-flow stirred tank reactor. J. Am. Chem. Soc. 111, 3870–3874 (1989).
Yoshida, R., Ichijo, H., Hakuta, T. & Yamaguchi, T. Self-oscillating swelling and deswelling of polymer gels. Macromol. Rapid Commun. 16, 305–310 (1995).
Yoshida, R., Yamaguchi, T. & Ichijo, H. Novel oscillating swelling-deswelling dynamic behaviour for pH-sensitive polymer gels. Mater. Sci. Eng. C 4, 107–113 (1996).
Yoshida, R., Takahashi, T., Yamaguchi, T. & Ichijo, H. Self-oscillating gel. J. Am. Chem. Soc. 118, 5134–5135 (1996).
Field, R. J. & Burger, M. (eds) Oscillations and Traveling Waves in Chemical Systems (John Wiley & Sons, New York, 1985).
Epstein, I. R. & Pojman, J. A. An Introduction to Nonlinear Chemical Dynamics: Oscillations, Waves, Patterns, and Chaos (Oxford University Press, New York, 1998).
Field, R. J., Körös, E. & Noyes, R. M. Oscillations in chemical systems. II. Through analysis of temporal oscillation in the bromate-cerium-malonic acid system. J. Am. Chem. Soc. 94, 8649–8664 (1972).
Amemiya, T., Ohmori, T. & Yamaguchi, T. An Oregonator-class model for photoinduced behavior in the Ru(bpy)32+-catalyzed Belousov-Zhabotinsky reaction. J. Phys. Chem. A 104, 336–344 (2000).
Yoshida, R. Self-oscillating gels driven by the Belousov-Zhabotinsky reaction as novel smart materials. Adv. Mater. 22, 3463–3483 (2010).
Yoshida, R., Tanaka, T., Onodera, S., Yamaguchi, T. & Kokufuta, E. In-phase synchronization of chemical and mechanical oscillations in self-oscillating gels. J. Phys. Chem. A 104, 7549–7555 (2000).
Maeda, S., Hara, Y., Yoshida, R. & Hashimoto, S. Peristaltic motion of polymer gels. Angew. Chem. Int. Ed. 47, 6690–6693 (2008).
Takeoka, Y., Watanabe, M. & Yoshida, R. Self-sustaining peristaltic motion on the surface of a porous gel. J. Am. Chem. Soc. 125, 13320–13321 (2003).
Sasaki, S., Koga, S., Yoshida, R. & Yamaguchi, T. Mechanical oscillation coupled with the Belousov-Zhabotinsky reaction in gel. Langmuir 19, 5595–5600 (2003).
Murase, Y., Maeda, S., Hashimoto, S. & Yoshida, R. Design of a mass transport surface utilizing peristaltic motion of a self-oscillating gel. Langmuir 25, 483–489 (2009).
Hara, Y. & Yoshida, R. Self-oscillating polymer fueled by organic acid. J. Phys. Chem. B 112, 8427–8429 (2008).
Hidaka, M. & Yoshida, R. Self-oscillating gel composed of thermosensitive polymer exhibiting higher LCST. J. Controlled Release 150, 171–176 (2011).
Suzuki, D., Kobayashi, T., Yoshida, R. & Hirai, T. Soft actuators of organized self-oscillating microgels. Soft Matter 8, 11447–11449 (2012).
Mitsunaga, R., Okeyoshi, K. & Yoshida, R. Design of comb-type self-oscillating gel. Chem. Comm. 49, 4935–4937 (2013).
Ito, Y., Nogawa, N. & Yoshida, R. Temperature control of the Belousov-Zhabotinsky reaction using a thermo-responsive polymer. Langmuir 19, 9577–9579 (2003).
Yoshida, R., Takei, K. & Yamaguchi, T. Self-beating motion of gels and modulation of oscillation rhythm synchronized with organic acid. Macromolecules 36, 1759–1761 (2003).
Shinohara, S., Seki, T., Sakai, T., Yoshida, R. & Takeoka, Y. Chemical and optical control of peristaltic actuator based on self-oscillating porous gel. Chem. Commun. 39, 4735–4737 (2008).
Shinohara, S., Seki, T., Sakai, T., Yoshida, R. & Takeoka, Y. Photoregulated wormlike motion of a gel. Angew. Chem. Int. Ed. 47, 9039–9043 (2008).
Yamamoto, T. & Yoshida, R. Self-oscillation of polymer and photo-regulation by introducing photochromic site to induce LCST changes. React. Func. Polym. 73, 945–950 (2013).
Tabata, O., Kojima, H., Kasatani, T., Isono, Y. & Yoshida, R. Chemo-mechanical actuator using self-oscillating gel for artificial cilia. Proceedings of the International Conference on MEMS 2003, pp 12–15 (2003).
Tabata, O., Hirasawa, H., Aoki, S., Yoshida, R. & Kokufuta, E. Ciliary motion actuator using self-oscillating gel. Sens. Actuat. A 95, 234–238 (2002).
Maeda, S., Hara, Y., Sakai, T., Yoshida, R. & Hashimoto, S. Self-walking gel. Adv. Mater. 19, 3480–3484 (2007).
Kuksenok, O., Yashin, V. V., Kinoshita, M., Sakai, T., Yoshida, R. & Balazs, A. C. Exploiting gradients in cross-link density to control the bending and self-propelled motion of active gels. J. Mater. Chem. 21, 8360–8371 (2011).
Yashin, V. V., Suzuki, S., Yoshida, R. & Balazs, A. C. Controlling the dynamic behavior of heterogeneous self-oscillating gels. J. Mater. Chem. 22, 13625–13636 (2012).
Murase, Y., Hidaka, M. & Yoshida, R. Self-driven gel conveyer: Autonomous transportation by peristaltic motion of self-oscillating gel. Sens. Actuat. B 149, 272–283 (2010).
Murase, Y., Takeshima, R. & Yoshida, R. Self-driven gel conveyer: effect of interactions between loaded cargo and self-oscillating gel surface. Macromol. Bioscience 11, 1713–1721 (2011).
Yoshida, R. & Murase, Y. Self-oscillating surface of gel for autonomous mass transport. Colloids Surf. B Biointerfaces 99, 60–66 (2012).
Shiraki, Y. & Yoshida, R. Autonomous intestine-like motion of tubular self-oscillating gel. Angew. Chem. Int. Ed. 51, 6112–6116 (2012).
Masuda, T., Hidaka, M., Murase, Y., Akimoto, A. M., Nagase, K., Okano, T. & Yoshida, R. Self-oscillating polymer brushes. Angew. Chem. Int. Ed. 52, 7468–7471 (2013).
Yoshida, R., Sakai, T., Ito, S. & Yamaguchi, T. Self-oscillation of polymer chains with rhythmical soluble-insoluble changes. J. Am. Chem. Soc. 124, 8095–8098 (2002).
Suzuki, D., Sakai, T. & Yoshida, R. Self-flocculating/self-dispersing oscillation of microgels. Angew. Chem. Int. Ed. 47, 917–920 (2008).
Suzuki, D. & Yoshida, R. Temporal control of self-oscillation for microgels by cross-linking network structure. Macromolecules 41, 5830–5838 (2008).
Suzuki, D. & Yoshida, R. Effect of initial substrate concentration of the Belousov-Zhabotinsky reaction on self-oscillation for microgel system. J. Phys. Chem. B 112, 12618–12624 (2008).
Suzuki, D. & Yoshida, R. Self-oscillating core/shell microgels. Polymer J. 42, 501–508 (2010).
Hara, Y. & Yoshida, R. A viscosity self-oscillation of polymer solution induced by the BZ reaction under acid-free condition. J. Chem. Phys. 128, 224904 (2008).
Suzuki, D., Taniguchi, H. & Yoshida, R. Autonomously oscillating viscosity in microgel dispersions. J. Am. Chem. Soc. 131, 12058–12059 (2009).
Taniguchi, H., Suzuki, D. & Yoshida, R. Characterization of autonomously oscillating viscosity induced by swelling/deswelling oscillation of the microgels. J. Phys. Chem. B 114, 2405–2410 (2010).
Ueno, T., Bundo, K., Akagi, Y., Sakai, T. & Yoshida, R. Autonomous viscosity oscillation by reversible complex formation of terpyridine-terminated poly(ethylene glycol) in the BZ reaction. Soft Matter 6, 6072–6074 (2010).
Ueki, T., Takasaki, Y., Bundo, K., Ueno, T., Sakai, T., Akagi, Y. & Yoshida, R. Autonomous viscosity oscillation via metallo-supramolecular terpyridine chemistry of branched poly(ethylene glycol) driven by the Belousov-Zhabotinsky reaction. Soft Matter 10, 1349–1355 (2014).
Ueki, T. & Yoshida, R. Self-oscillating micelles. Chem. Comm. 49, 6947–6949 (2013).
Dayal, P., Kuksenok, O. & Balazs, A. C. Designing autonomously motile gels that follow complex paths. Soft Matter 6, 768–773 (2010).
Dayal, P., Kuksenok, O. & Balazs, A. C. Using light guide the self-sustained motion of active gels. Langmiur 25, 4298–4301 (2009).
Kuksenok, O. & Balazs, A. C. Modeling the photoinduced reconfiguration and directed motion of polymer gels. Adv. Funct. Mater. 23, 4601–4610 (2013).
Yashin, V. V. & Balazs, A. C. Pattern formation and shape changes in self-oscillating polymer gels. Science 314, 798–801 (2006).
Yashin, V. V., Kuksenok, O. & Balazs, A. C. Modeling autonomously oscillating chemo-responsive gels. Prog. Polym. Sci. 35, 155–173 (2010).
Yashin, V. V., Kuksenok, O., Dayal, P. & Balazs, A. C. Mechano-chemical oscillations and waves in reactive gels. Rep. Prog. Phys. 75, 066601 (2012).
Yashin, V. V., Kuksenok, O. & Balazs, A. C. Computational design of active, self-reinforcing gels. J. Phys. Chem. B 114, 6316–6322 (2010).
Lohmeijer, B. G. G. & Schubert, U. S. Supramolecular engineering with macromolecules: An alternative concept for block copolymers. Angew. Chem. Int. Ed. 41, 3825–3829 (2002).
Ueki, T., Watanabe, M. & Yoshida, R. Belousov-Zhabotinsky reaction in protic ionic liquids. Angew. Chem. Int. Ed. 51, 11991–11994 (2012).
Zhou, H., Ding, X., Zheng, Z. & Peng, Y. Self-regulated intelligent systems: where adaptive entities meet chemical oscillators. Soft Matter 9, 4956–4968 (2013).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Yoshida, R., Ueki, T. Evolution of self-oscillating polymer gels as autonomous polymer systems. NPG Asia Mater 6, e107 (2014). https://doi.org/10.1038/am.2014.32
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/am.2014.32
Keywords
This article is cited by
-
Self-oscillating chemoelectrical interface of solution-gated ion-sensitive field-effect transistor based on Belousov–Zhabotinsky reaction
Scientific Reports (2022)
-
Creation of softmaterials based on self-oscillating polymer gels
Polymer Journal (2022)
-
Moisture-induced autonomous surface potential oscillations for energy harvesting
Nature Communications (2021)
-
Harnessing the power of chemically active sheets in solution
Nature Reviews Physics (2021)
-
A programmable chemical computer with memory and pattern recognition
Nature Communications (2020)
